Article Text

Abstract
Background—Offspring with a family history of Crohn’s disease have an earlier age of onset than their parents. This might be due to genetic anticipation, characterised by earlier and/or more severe disease in subsequent generations.
Aims—To investigate the possibility of genetic anticipation in affected parent-child pairs with Crohn’s disease from France and Belgium.
Patients and methods—In a cohort of 160 multiply affected families with Crohn’s disease, 57 parent-first affected child pairs were detected. Clinical characteristics (age at diagnosis, disease extent, and type) of both parents and children were registered and compared.
Results—Children were younger than their parents at diagnosis in 48/57 (84%) pairs. The median age at diagnosis was 16 years younger in children than in parents (p<0.0001). However, the difference was related to the age at diagnosis in the parents and was not present in 12 parent-child pairs with an early age at diagnosis for the parents. In most cases, disease extent and type were not considered more severe in children than in parents. Parental sex affected neither age at diagnosis nor extent and type of disease in children.
Conclusion—Patients in the second affected generation acquire their disease at an earlier time in life in some but not all familial cases of Crohn’s disease. Several explanations including genetic anticipation and environmental factors might explain this phenomenon.
- Crohn’s disease
- familial
- genetic anticipation
Statistics from Altmetric.com
A recent survey of clinical characteristics in 72 multiply affected families with Crohn’s disease (CD) showed that age at onset of illness was significantly lower in patients with a family history of CD than in those with no such history (22 years versus 26.5 years).1 In parent-child pairs, parents had a significantly higher median age at onset or diagnosis than offspring.2-4 Genetic anticipation has been suggested as a possible explanation for this finding.2
The term anticipation denotes an increase in severity or decrease in the age at onset as a disease is passed through generations. It has been described in monogenic neurological illnesses such as Huntington’s disease, myotonic dystrophy, and more recently Friedreich’s ataxia.5-7 In these diseases, there is firm evidence that anticipation reflects the effects of genetic factors and that it has a true molecular basis. Amplification of DNA triplet repeats within or adjacent to the disease gene occurs in successive generations. This DNA instability is associated with increasing disease severity and earlier age at onset.8-11 In Friedreich’s ataxia, the number of GAA repeats within the frataxin gene is strongly correlated with age at onset and with the rate of disease progression, suggesting that the expansion itself is the cause of the disease.12 Another phenomenon, genomic imprinting, has been described in association with genetic anticipation in several diseases. Imprinting is observed when affected offspring show different phenotypic effects of genes carried on maternally derived as opposed to paternally derived chromosomes.13 It may induce differences in age of onset, disease type, or severity.
The list of conditions exhibiting anticipation is growing rapidly,14 and recognising anticipation and imprinting would be very important for a proper understanding of inherited susceptibility in CD. For instance, if genetic anticipation exists in CD it might be reasonable, when performing linkage analysis, to separate families with and without the anticipation pattern in an attempt to address genetic heterogeneity. Preliminary evidence for genetic anticipation in CD has been proposed by the Johns Hopkins group in Baltimore, Maryland,15 and, very recently, CAG repeat expansions have been observed in a subset of families with CD.16 However, epidemiological data for genetic anticipation are subject to many biases, and large numbers of families are needed to assess it properly.14 ,17
The aim of this study was to look for genetic anticipation in affected parent-child pairs with CD, most of them from a limited geographical area in northern France and Belgium.
Patients and methods
STUDY SAMPLE
One hundred and sixty families in whom two or more first degree relatives have inflammatory bowel disease were recorded between 1956 and 1995 in northern France, through the GETAID (France) and in Leuven and Liège (Belgium). In 57 families, at least one parent and one child had CD. These families included 44 with one parent and one child affected. In 13 families more than two members were affected—one parent and more than one child. Eight families had two, four had three, and one had five affected children. In order not to use some data points more than once, only the 57 parent-first affected child pairs were considered in this study. Of the affected parents, 21 were male and 36 female. Of the offspring, 22 were male and 35 female. Thirty one families were identified in Leuven, 12 in Lille, nine in Liège, and five through the GETAID. Forty nine per cent of the families were identified from university hospital records and 51% were identified in population based studies through the Registre des Maladies Inflammatoires du Tube Digestif du Nord-Ouest de la France (EPIMAD) and the Flemish Crohn’s and Colitis Patients Association. The diagnostic criteria for CD were those previously described.18 The study, which was not cross sectional, was part of a larger collaborative research programme on genetics of inflammatory bowel disease (IBD) in which all families recorded in the different centres were included.
CLINICAL DATA
A standardised questionnaire on family history and clinical details for each patient was used in all participating centres. Family history included a pedigree, year of birth, and sex of all first degree relatives. The following clinical characteristics were collected for each patient with CD: age at diagnosis, disease location at diagnosis (small bowel only, colon only, small bowel and colon), and disease type—primarily fibrostenotic (cicatrising), primarily inflammatory, or primarily perforating (fistulising).19 Type of disease was inferred from the dominant clinical and morphological features: in the fibrostenotic form, patients had clinical and radiological evidence of stenosis; in the fistulising type, patients had acute free perforation, subacute abscess formation, or chronic internal fistulae; patients with the inflammatory form had no evidence of stenosis or fistulae. Age at onset (first symptoms) was also recorded when available. Clinical data were obtained from 134 patients with CD (54 males, 80 females); median duration of disease was 12.5 years (range 7–20). All the data were rechecked by one senior investigator in each centre at the time of the study.
METHODS
To test the families for anticipation, the differences in age at diagnosis (δ) and disease severity at diagnosis were recorded for parent-child pairs. By definition, disease severity was based on the extent of disease and the type of the disease. Disease extent was considered greater when both small bowel and colon were involved as compared with small bowel or colon only. Other cases were considered equal. With respect to disease type, the primarily perforating type was considered to be more severe than the primarily inflammatory or fibrostenotic types.
To search for evidence of parental awareness, time between onset of symptoms and diagnosis was compared in parents and children in 19 families in whom this information was available.
Possible imprinting was evaluated by dividing the sample into two groups, one with affected fathers and the other with affected mothers.
STATISTICS
Matched analysis was used for the pairs. Statistical analysis of δ was calculated using the Wilcoxon signed rank test. A value of p<0.05 was considered significant. Median and interquartile ranges (Q1–Q3) were used for summarising statistical central and dispersion measures.
Results
AGE AT DIAGNOSIS
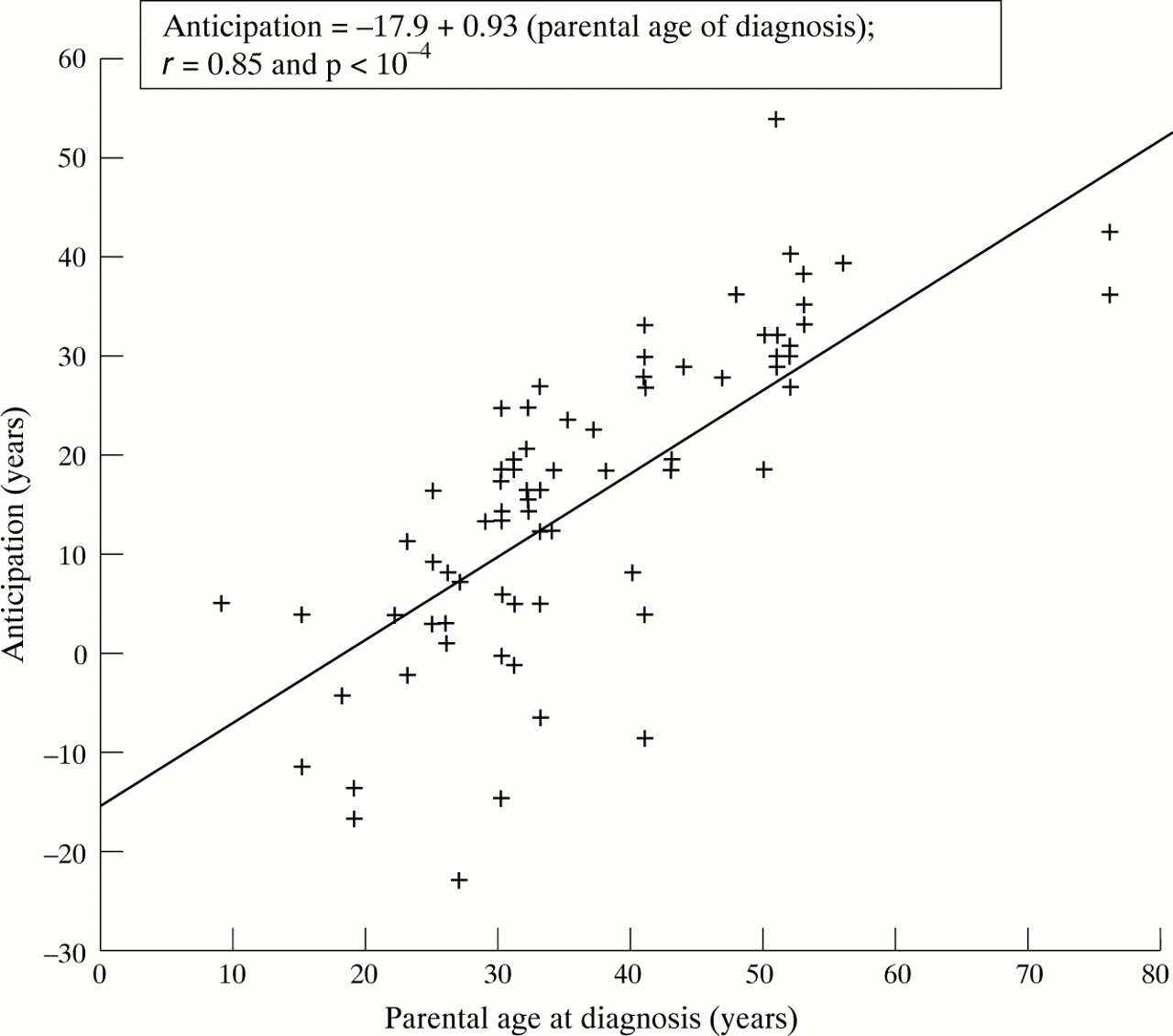
The median age at diagnosis was 33 years (range 29–48) in the parents and 20.5 years (range 15.5–27) in the children. The median difference in age at diagnosis between parents and children (δ) was 16 years (range 4–26) (p<0.0001). Children were younger than their parents at diagnosis in 48/57 (84%) pairs (table 1). These data are in agreement with anticipation for age at diagnosis. There was a significant correlation between parental age at diagnosis and anticipation (r=0.85; p<0.001) (fig 1).
Indicators of anticipation in 57 parent–child pairs

Correlation between parental age at diagnosis and anticipation.
The calendar years at which the diagnosis was made were discordant for parents (median 1978, range 1965–1984) and their offspring (median 1986, range 1982–1992). In six of the 57 pairs, the parent was diagnosed after the child.
Epidemiological data from the Register of IBD of northern France indicated a median age of diagnosis for sporadic non-familial CD of 26.5 years.18 In our sample, 12/57 parents (21%) were defined as having an early diagnosis (at age less than 26.5 years). When considering parent-child pairs in which the parents had CD at an early age (less than 26.5 years), δ dropped to two years (not significant). There was a significant difference between the number of pairs showing anticipation among “early diagnosed” parent-child pairs and “late diagnosed” (greater than 26.5 years) parent-child pairs (table 1). Twenty five per cent of the parents had their child after their diagnosis of CD. This percentage was significantly higher in early diagnosed pairs (83%) than in late diagnosed pairs (11%) (p<0.0001).
DISEASE SEVERITY
In fig 2 disease extent within pairs is compared. Positive anticipation according to extent—that is, extent of disease greater in the younger than in the older generation, was observed in 12/57 (21%) pairs while 36/57 pairs (63%) showed a similar extent of disease.

Comparison of Crohn’s disease extent in 57 parent-child pairs.
Figure 3 shows the comparison of disease type within pairs. Disease type was considered more severe in 2/57 pairs (3%). It was similar in 81% of pairs.

{kind=link}
{kind=link}
{kind=link}
Comparison of Crohn’s disease type in 57 parent-child pairs. Primarily perforating type was considered more severe than primarily inflammatory and fibrostenotic types.
PARENTAL AWARENESS
Age at onset of symptoms was recorded in 19 pairs. Time between onset of symptoms and diagnosis was not significantly shorter in children than in their parents. Mean difference was 1.8 years.
Discussion
We confirmed in this large series that there is an important generational difference in age at diagnosis in familial CD. Age of diagnosis of the offspring was younger at diagnosis in 48/57 pairs (84%) with a median difference of 16 years.
Similar observations have recently been presented in the USA and UK. Polito et al identified 27 pairs of two generation first degree relatives treated for CD at Johns Hopkins Hospital and 32 through a multicentre survey in the USA.15 Age at diagnosis was earlier in the younger member in 23/27 (85%) pairs and 31/32 pairs (97%) respectively. A study in the UK collected 77 pairs of which 79% of the parents were more than five years older than the child at onset (more than 10 years in 60.4%).3Interestingly, the parent-child difference in age was very similar in these two studies and in agreement with our findings.
In view of this consistent finding, a hypothesis worthy of consideration is genetic anticipation.15 However, there are difficulties in defining biological anticipation.20 As discussed in depth with respect to several neurological or other diseases,14 ,17 ,21-23 the epidemiological data supporting genetic anticipation can be subject to many biases, particularly in retrospective studies. Firstly, an increased awareness and diagnostic acuity may influence age at diagnosis in the later generation. Earlier diagnosis in the children appeared not to be related to increased parental awareness since it only accounted for less than two years. Diagnostic facilities have improved between the 1960s and the 1980s: routine colonoscopy was introduced between 1975 and 1980 in our region. It is unlikely however that the difference in age at diagnosis, which averaged 16 years, is a reflection of the improvements in diagnostic testing. The design of all of these studies might have introduced a first ascertainment bias—that is, the members of the later affected generation have not yet lived through their later years of susceptibility.24 ,25 This could be especially true if one considers that several studies have shown a second peak of susceptibility for CD at age 50.26 This latter was not the case in our epidemiological register.18 More importantly, if genetic anticipation exists, it should be independent of the parents age at diagnosis. However, as illustrated in fig 1, there was a good correlation between parental age at diagnosis and the degree of anticipation. The main bias when looking for genetic anticipation, is probably the preferential ascertainment of parents with late age at diagnosis. The median age at diagnosis in the parents was 33 years in the present families, and 31.4 years and 33.1 years in the two series from Johns Hopkins Hospital15; the age at onset was 38 years in Oxford.3 All these figures are higher than the ones observed in sporadic cases.2 ,26 It might reflect a selection bias with less recruitment of parents with an early onset and reduced fertility.27 Accordingly, only 25% of children were born after the diagnosis of CD in their parents. In order to control this bias, we stratified parent-child pairs according to age at diagnosis in the parents. No anticipation was present in the early diagnosed parent-child pairs. Eighty three per cent of the children from these pairs were born after the diagnosis of CD in their parents. This difference could also suggest genetic heterogeneity in CD, with a subtype exhibiting anticipation. It is also worth noting that in the recent molecular study of IBD families by Cho et al,16 the child had a longer (CAG)n repeat than the parent in only 6/14 CD-CD pairs.
The concept of genetic anticipation encompasses an increased severity of the disease in the member of the later generation. Although there is so far no accepted index of severity in CD, it can be approached using disease extent and type. When considering these two citeria (figs 2 and3), we found no evidence of increased severity of CD in children when compared with their parents. Similarly, Satsangi et alreported that parent and child were concordant for extent in 75% of 31 CD-CD pairs.3 Conversely, Polito et al found a greater extent of CD in 15 of their 27 pairs.15 These differences might reflect the source of their patients. In the latter study, patients were solely recruited at Johns Hopkins Hospital, a reference centre where more severely ill patients are seen. More generally, the relevance of CD phenotypes as markers of anticipation needs to be discussed. Clinical data were checked at the time of the study and an effort was made to standardise appreciation of disease extent and type between centres. However, the patients were primarily investigated by a large number of clinicians and it is inevitable that different investigations were used. Clinical characteristics were evaluated at diagnosis. Although there is evidence that extent and severity of CD may be dynamic, changing over time, several studies have stressed the importance of initial disease location as a predictor of the clinical pattern that patients might expect.28-30 The point of view that fistulising is more severe than other types has received support from several studies,31-33 but controversial results have been reported.34 ,35 Finally, the classification of CD into primary perforating, inflammatory, or fibrostenotic disease has yet to be validated in a prospective fashion.
Imprinting has been described in association with genetic anticipation in several diseases.13 Recognition of imprinting within pedigrees is simpler than recognition of anticipation. No major ascertainment bias can interfere. In this study we observed no influence of parental sex on age at diagnosis, disease extent, or type of CD in children.
In summary, it appears that patients in the second affected generation acquire their disease at an earlier time in life in familial CD. So far, we could not conclude that there is a genetic cause for this. Ascertainment bias cannot be excluded in this and other retrospective studies. Furthermore, alternative explanations for the consistent spread in age at onset between parents and children can be proposed. A cohort effect due to simultaneous exposure to an environmental pathogen was not apparent: the calendar year of diagnosis in parent and child in this study and the one from Satsangi et al 3were discordant. As noted by Sachar,36 an increased exposure to an environmental agent “to which people are exposed at progressively younger ages and higher concentrations” over time would also result in disease at younger ages in the children of the parent-child pairs.
The optimal design for a study of anticipation would be to consider only prospectively obtained cases.14 Because of the rarity of familial CD, this seems impracticable. Further studies of a very large dataset of multiply affected families with standardised methodology and minimising biases of ascertainment are now required. Molecular understanding of the genetics of CD will also help to resolve this question.37-39
Acknowledgments
This work was supported partly by the Association F. Aupetit, the Ministère de la Santé et de l’Action Humanitaire (Direction Générale de la Santé), INSERM (Grant 92/R/2), CH et U de Lille, and the companies Ferring and Astra. We thank the GETAID and in particular, R Modigliani, J P Gendre, and J L Dupas who contributed to the study, and the Flemish Crohn and Colitis Association.