Article Text

Abstract
Background/aims—The gene promoter for the intercellular adhesion molecule ICAM-1 possesses binding sites for several transcriptional factors, including nuclear factor κB (NF-κB). The role of NF-κB in ICAM-1 gene regulation was therefore examined by using different proteasome inhibitors in tumour necrosis factor α (TNF-α) stimulated IEC-6 rat intestinal epithelial cells.
Methods—ICAM-1 expression was analysed by enzyme linked immunosorbent assay (ELISA), reverse transcriptase polymerase chain reaction, and immunohistochemistry. Steady state levels of cytoplasmic IκB protein were evaluated by western blot, and nuclear translocation of NF-κB was determined by electrophoretic mobility shift assay and immunofluorescence staining. Cell adhesion was assayed by measuring the binding of fluorescence labelled MOLT-4 cells.
Results—TNF-α induced ICAM-1 mRNA and protein expression in IEC-6 cells, which was followed by increased adhesion of MOLT-4 lymphocytes. Blocking TNF-α induced IκBα degradation with proteasome inhibitors reduced TNF-α induced NF-κB activation and ICAM-1 gene induction and notably decreased MOLT-4 cell adhesion without affecting Jun N-terminal kinase (JNK/SAPK) activity or de novo protein synthesis.
Conclusion—TNF-α induction of ICAM-1 expression is mediated by the transcription factor NF-κB and can be inhibited by blocking IκBα degradation. Thus the IκB/NF-κB system is a promising target for pharmacological modulation of the expression of adhesion molecules and other inflammatory genes in the intestine.
- adhesion molecules
- ICAM-1
- cytokines
- tumour necrosis factor α
- intestinal inflammation
- NF-κB.
Statistics from Altmetric.com
Infiltration and persistence of inflammatory cells within tissues are hallmarks of inflammatory disorders including ulcerative colitis and Crohn’s disease.1 Recruitment and activation of these cells to the inflammatory focus are multifactorial events involving adhesion of circulating cells to the vascular endothelium followed by tissue migration. Cell to cell and cell to matrix interactions are critical elements in the inflammatory and immune response and are dependent on coordinated expression of various adhesion molecules.2 The intercellular adhesion molecule-1 (ICAM-1) is an active participant in mediating leucocyte adhesion to endothelial and epithelial cells.2 ICAM-1 (CD54) is a cell surface glycoprotein that belongs to the immunoglobulin superfamily of adhesion molecules2 and binds to the β2-integrin counter receptors LFA-1 (CD11a/CD18) and MAC-1 (CD11b/CD18). Basal levels of membrane ICAM-1 protein are detected on several human tumour intestinal epithelial cell (IEC) lines; this molecule is up regulated by various proinflammatory cytokines including tumour necrosis factor α (TNF-α) and interferon γ.3-6 Recently, ICAM-1 expression on transformed human IECs has been shown to be enhanced by bacterial or cytokine stimulation,6 and systemic administration of lipopolysaccharide up regulated small intestinal ICAM-1 by threefold,7 although the location of the enhanced expression was not determined. Expression of ICAM-1 by IECs may represent a critical step in regulating interaction between lamina propria lymphocytes, neutrophils, and IECs. Many cellular functions involve interaction of ICAM-1 with MAC-1 and LFA-1. For example, the LFA-1/ICAM-1 pathway is involved in MHC restricted antigen presentation,8 cell proliferation triggered by antigens and mitogens,9 ,10 and T cell mediated cytotoxicity.11 Also, blockade of ICAM-1 by neutralising antibodies inhibits the adherence of neutrophils to intestinal epithelial cell monolayers,6 ,12 and in a small preliminary study in a single institution, ICAM-1 antisense oligonucleotide treatment was reported to have some benefit in steroid dependent Crohn’s disease, although toxicity effects must be carefully analysed.13 Therefore, it is important to understand the regulation of ICAM-1 gene expression in IECs after cytokine stimulation to help guide the rational development of pharmacological modulators of this key adhesion molecule.
Although the ICAM-1 gene promoter has binding sites for many transcription factors, including SP-1, C/EBPb/NF-IL-6 and nuclear factor κB (NF-κB), mutational analysis showed that the presence of NF-κB binding sites is critical for ICAM-1 promoter activity.14 ,15 The transcription factor NF-κB is involved in the rapid induction of a number of cytokines and adhesion molecules implicated in the immune and inflammatory response.16 ,17 This factor is formed by several different dimers of members of the Rel family, which include NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel.17 The heterodimers composed of the subunits NF-κB1 and RelA are the most frequent and active forms of NF-κB.18 In most resting cells, NF-κB is found in the cytoplasm associated with inhibitory molecules called IκBs.19 NF-κB/IκB complexes are sequestered in the cytoplasm, thereby inhibiting NF-κB dependent gene expression. Appropriate stimuli, including interleukin (IL)-1 and TNF-α,20 ,21 ubiquinate a kinase which phosphorylates IκBα at serine residues 32 and 36.22 Phosphorylated IκBα is then selectively ubiquinated and rapidly degraded via a non-lysosomal ATP dependent 26S proteolytic complex composed of a 700 kDa proteasome.23-25 The free NF-κB is then translocated to the nucleus and induces genes that have κB binding sites, including proinflammatory cytokines, class II MHC antigens, and adhesion molecules including ICAM-1. Importantly, NF-κB controls the expression of its own inhibitor.26 Activation of NF-κB induces the IκBα gene, leading to a rapid replenishment of IκB protein, which again complexes with NF-κB and thus down regulates the activation process.26
We have recently shown that NF-κB regulates cytokine induced IL-8 expression in native and transformed IECs and that blockade of IκBα degradation inhibits IL-8 production.27 In addition, we have also demonstrated the feasibility of down regulating proinflammatory gene expression by adenoviral delivery of an NF-κB super-repressor in IECs.28 Consequently, IκB represents a potential target for pharmacological therapy of intestinal inflammation. In the present study, we examined the signalling pathways associated with TNF-α induction of ICAM-1 gene expression in the non-transformed rat intestinal epithelial cell line IEC-6 and investigated the role of the IκB/NF-κB system in this process by inhibiting IκBα degradation with two different proteasome inhibitors: N-acetyl-Leu-Leu-Nle (ALLN) and Z-Leu-Leu-Leu-H (MG-132). ALLN is a cysteine protease inhibitor that blocks calpain 1 activity, but is also capable of inhibiting the chymotrypsin-like activity of the proteasome complex when used at relatively high doses (100–200 mM).23 ,24 ,29-33 The peptide aldehyde MG-132 is a more potent and specific proteasome inhibitor than ALLN.22-24 29-34
Methods
CELL CULTURE
The rat non-transformed intestinal epithelial cell line IEC-6 (ATCC CRL 1592) was used between passage 3 and 15 and was grown in high glucose Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, New York, USA) with 5% heat inactivated fetal bovine serum (Gibco), 2 mMl-glutamine, 1 U/ml insulin, and antibiotics (Pen/Strep/Fungisone, 1 ×; Gibco). Cells were cultured in a water saturated atmosphere of 95% air and 5% CO2. For protein or mRNA analysis, 3 × 105 or 5 × 105 cells respectively were seeded into a six-well tissue culture plate (2 ml/well) (Costar, Cambridge, Massachusetts, USA) and grown to 50–70% confluency (normally 1–2 days after seeding). The medium was removed and the cells were preincubated with ALLN (50 μg/ml; Boehringer Mannheim, Indianapolis, Indiana, USA) or MG-132 (5 μg/ml; Peptide International, Louisville, Kentucky, USA) for 30 minutes, after which they were stimulated with rat recombinant TNF-α (10 ng/ml; Intergen, Purchase, New York, USA). Cell viability after exposure to ALLN was checked by the trypan blue dye exclusion test and was found to be greater than 90%.
METABOLIC LABELLING OF CELLS
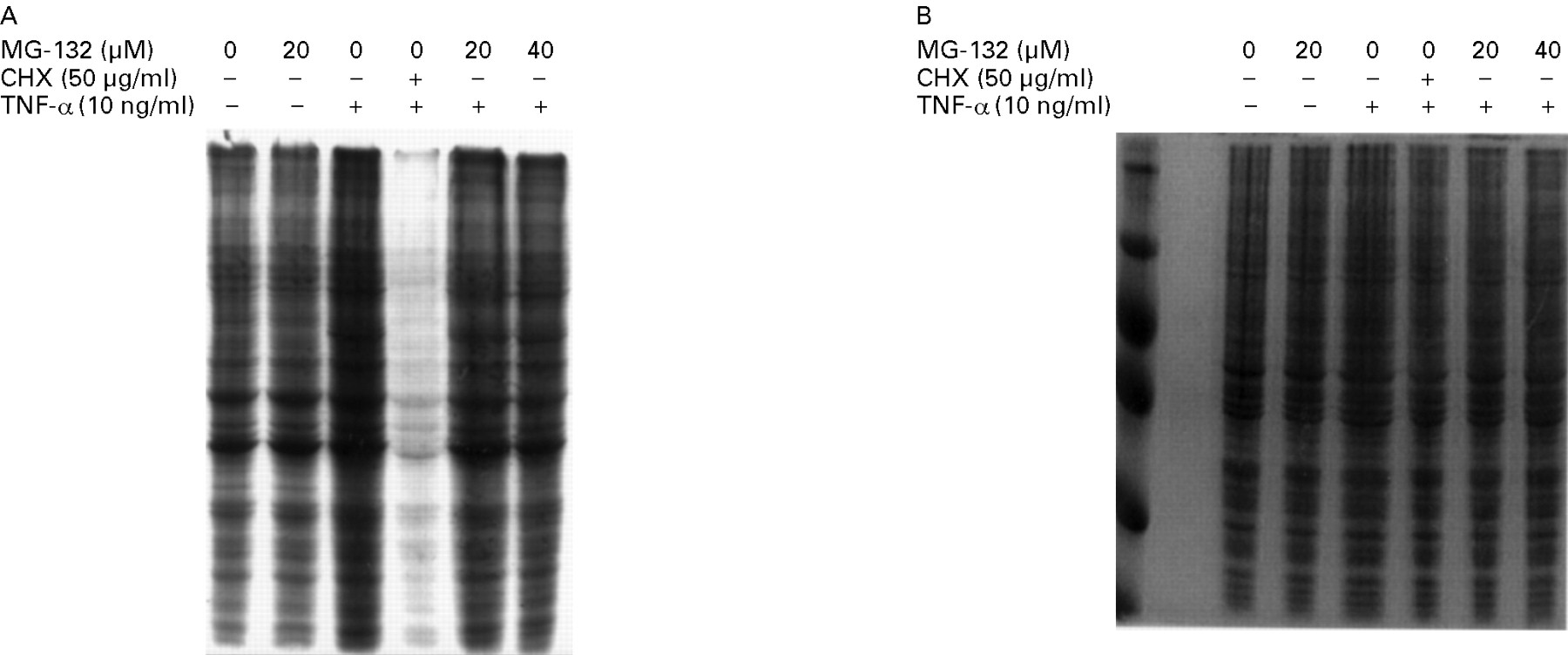
IEC-6 cells were cultured in medium containing 150 μCi [35S]methionine (1000 Ci/mmol; ICN Pharmaceuticals, Costa Mesa, California, USA) for 12 hours with or without various doses of MG-132 (20–40 μM) in the presence or absence of TNF-α (10 ng/ml). Cycloheximide (50 μg/ml) was used as a positive control of protein synthesis inhibition. At the end of the incubation period, cells were lysed in 1 × Laemmli buffer, and 20 μg protein was electrophoresed on sodium dodecyl sulphate (SDS)/10% polyacrylamide gel. The gels were stained with Coomassie blue and then impregnated with salicylate and exposed as described.35
RNA EXTRACTION AND AMPLIFICATION BY REVERSE TRANSCRIPTASE POLYMERASE CHAIN REACTION (RT-PCR)
Cells (3 × 105/well in six-well tissue culture plates; Costar) were pretreated with ALLN (50 μg/ml) for 30 minutes and then stimulated with 10 ng/ml TNF-α for four hours. RNA was isolated using the Trizol method (Gibco) according to the manufacturer’s specifications. The integrity and quality of each sample was checked by staining with ethidium bromide. First strand cDNA was synthesised using 1 μg total RNA, 15 U RNA guard (Pharmacia, Piscataway, New Jersey, USA), 1 × first strand buffer (Gibco), 12.5 mM dNTP (Pharmacia), 125 pmol random hexamer primers (Pharmacia), and 125 U Moloney murine leukaemia virus reverse transcriptase (Gibco) in a final volume of 25 μl. The reaction was carried out for one hour at 39°C followed by seven minutes at 93°C, one minute at 24°C, and then slowly cooled down at 4°C for 20 minutes.36 PCR was carried out in a volume of 50 μl containing 5 μl of the reverse transcription mixture, 1 × Taq buffer (Applied Biosystems, Foster City, California, USA), 0.4 μM each primer, 200 μM dNTPs, and 2.5 UThermo aquaticus poly merase (Applied Biosystems). PCR was carried out in a 9600 Perkin-Elmer cycler (Applied Biosystems) for 25 to 35 cycles to monitor the linearity of the amplification. The ICAM-1 oligonucleotide primers were: (5′) 5′-GATGCTGACCCTGGAGAGCA-3′ and (3′) 5′-AGCACTTGCGGTCCACGATG-3′. The actin oligonucleotide primers were: (5′) 5′-ACCACAGCTGACAGGGAAATCG -3′ and (3′) 5′-AGAGGTCTTTACGCAT GTCAACG-3′). The lengths of the amplified products were 409 and 280 bp respectively. The PCR temperatures used were 94°C for 45 seconds, 55°C for 30 seconds, and 72°C for 90 seconds followed by an extension of 10 minutes at 72°C. The PCR products (10 μl) were electrophoresed on a 2% agarose gel containing ethidium bromide. A negative from the photographs of the gels taken with a Polaroid 665 film (Polaroid Corp., Cambridge, Massachusetts, USA) was used and scanned with a Silverscanner II PS v2.1a connected to a Power Mac 8100/80 computer and analysed with Adobe Photoshop 2.5.1 software. Negative controls consisted of tubes with (a) no nucleic acid or (b) RNA only.
NUCLEAR EXTRACTS
Cells were plated (2 × 106 cells) in 100 mm diameter dishes (Costar) and stimulated for 30 minutes with TNF-α (10 ng/ml). They were then lysed and nuclear protein was prepared as described previously.27
ELECTROPHORETIC MOBILITY SHIFT ASSAY (EMSA)
Nuclear extracts (2 μg) were incubated with double stranded class 1 MHC κB sites (GGCTGGGGATTCCCCATCT), separated by electrophoresis, and analysed by autoradiography as described previously.27 For antibody supershift analysis, nuclear extracts were preincubated with 1 μl RelA antibody directed against the C-terminal portion of the molecule (Rockland, Gilbertsville, Pennsylvania, USA) or with 1 μl p50 antibody directed against the nuclear localisation signal portion of the molecule (SC-144X; Santa Cruz Biotechnology; Santa Cruz, CA, USA) for 15 minutes at room temperature before the addition of the binding buffer and probe. Blocking with a non-degradable IκBα delivered by an adenoviral vector (Ad5IκB) was performed as described.28
WESTERN BLOT ANALYSIS
Cells (3 × 105/well in six-well tissue culture plates; Costar) were pretreated with ALLN (50 μg/ml) or vehicle for 30 minutes and then stimulated with 10 ng/ml TNF-α for various times. After removal of the medium, the cells were lysed in 1 × Laemmli buffer, and 20 μg protein was electrophoresed on SDS/10% polyacrylamide gels. Immunoreactive IκBα was detected using the enhanced chemiluminescence light (ECL) detecting kit (Amersham, Arlington Heights, Illinois, USA) as described previously.27
IMMUNOFLUORESCENT DETECTION OF RelA (p65)
Cells (1 × 105/35 mm diameter dish; Costar) were preincubated with ALLN (50 μg/ml) or MG-132 (5 μg/ml) for 30 minutes and then stimulated for 30 minutes with TNF-α (10 ng/ml). They were then fixed with 100% ice cold methanol. Blocking was performed using 25% non-immune goat serum (Sigma, St Louis, Missouri, USA) for 30 minutes. Rabbit anti-RelA antibody (diluted 1:200 in 25% non-immune goat serum) was then added for 30 minutes, after which rhodamine isothiocyanate conjugated goat anti-rabbit IgG antibody (Jackson ImmunoResearch, West Grove, Pennsylvania, USA) diluted 1:100 in 25% non-immune goat serum was added for 30 minutes. RelA expression was visualised with a fluorescent light microscope.
LYMPHOCYTE BINDING ASSAY
IEC-6 cells were seeded into 24-well plates. After they had reached about 70% confluency, the medium was changed and some wells were preincubated with ALLN (50 μg/ml) or aprotinin (10 μg/ml; Boehringer Mannheim) for 30 minutes before the addition of TNF-α (10 ng/ml). After an additional 16 hours of incubation, lymphocyte binding was determined as previously described.36 Briefly, MOLT-4 cells (ATTC CRL 1582), a human lymphocyte cell line, were fluorescently labelled with 2,7-bis-(2-carboxyethyl)-5- (and -6-) carboxyfluorescein acetoxymethyl ester (BCECF-AM) (Molecular Probes, Eugene, Oregon, USA). The labelled MOLT-4 cells (106/well) were incubated with the cultured IEC-6 cells for one hour at 37°C in RPMI medium without serum. After three washes with RPMI medium, phosphate buffered saline (PBS) containing 2% Nonidet P40 was added (500 μl/well), and the mixture transferred to a cuvette. The samples were excited at 485 nm, and the emission was measured at 530 nm.
ENZYME LINKED IMMUNOSORBENT ASSAY (ELISA) OF ICAM-1
IEC-6 cells were seeded into 96-well plates and grown until about 70% confluency was obtained. The medium was changed and cells were incubated for 16 hours with TNF-α (10 μg/ml), some with 30 minutes preincubation with ALLN (50 μg/ml) or aprotinin (10 μg/ml). Afterwards, an ELISA for ICAM-1 was performed as previously described.36 Briefly, cells were fixed with 1% paraformaldehyde, blocked with 0.25% gelatin. The first antibody was a mouse anti-rat ICAM-1 antibody (Seikagaku America, Rockville, Maryland, USA), diluted 1:100, and the secondary antibody was a goat anti-mouse alkaline phosphatase conjugated antibody (Santa Cruz), diluted 1:200.
IMMUNOHISTOCHEMICAL ANALYSIS OF ICAM-1 EXPRESSION
IEC-6 cells were grown on glass coverslips in six-well plates. After stimulation of some wells with TNF-α for 16 hours, cells were fixed with 100% ice cold methanol for 10 minutes. After two washes with PBS, blocking was performed with 1% goat serum diluted in PBS for 30 minutes at room temperature. The slides were incubated for one hour at room temperature with either mouse anti-rat ICAM-1 antibody diluted 1:100 in PBS or non-specific mouse IgG (diluted to the same protein concentration as the mouse anti-rat ICAM-1 antibody in PBS) as a negative control. Subsequent incubations with biotinylated secondary antibody and a preformed avidin/biotinylated enzyme complex were performed with the Vectastain ABC kit (Vector Laboratories, Burlingame, California, USA) according to the manufacturer’s instructions. Colour was developed by incubating the slides for 5–10 minutes with diaminobenzidine (DAB) using the fast-DAB tabs (Sigma Chemical Co). Counterstaining was performed with haematoxylin according to standard protocols.
WHOLE CELL EXTRACTS
Cells were plated (2 × 106) in 100 mm dishes and grown until about 80% confluency was obtained. The cells were washed with PBS and then grown under “serum starved” conditions (0.5% fetal bovine serum) for 48 hours and finally treated with TNF-α (10 ng/ml) or TNF-α plus ALLN (50 μg/ml) for 30 minutes. The cells were scraped with a rubber policeman, washed with ice cold PBS, and then lysed in Dignam C buffer (420 mM NaCl, 1.5 mM MgCl2, 20 mM Hepes, pH 7.0, 0.2 mM EDTA, 25% glycerol, 0.5 mM dithiothreitol) containing protease and phosphatase inhibitors (0.5 mM phenylmethanesulphonyl fluoride, 0.1 mM p-nitrophenyl phosphate, 0.04 mM β-glycerophosphate, 0.05 mM Na3VO4, 40 mg/ml bestatin, 2 mg/ml aprotinin, 0.54 mg/ml leupeptin, 0.7mg/ml pepstatin A). Lysates were rotated at 4°C for 30 minutes. Cell membranes were then pelleted by cold centrifugation at 14 000 rpm and discarded. The supernatant was divided into aliquots and stored at −80°C. Protein concentration of whole cell extracts was determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, California, USA).
ASSAY OF c-Jun N-TERMINAL KINASE (JNK/SAPK)
JNK/SAPK activity was assessed in IEC-6 cells using an in vitro kinase assay as described previously.37 Recombinant glutathione S-transferase (GST)-c-Jun protein (amino acids 1–79) containing the activation domain of c-Jun, a mutated GST-c-Jun with Ser63 and Ser73 mutated to Ala (GST-c-JunAA), or GST protein alone were utilised as substrates. A 25 μg portion of whole cell extract was incubated with 5 μg substrate protein linked to glutathione-Sepharose beads. After extensive washing of the complexes, the kinase reaction was performed with [γ-32P]ATP (4500 Ci/mmol; ICN Pharmaceuticals). The proteins were fractionated using SDS/polyacrylamide gel electrophoresis (PAGE) (12.5% gel) and visualised/quantified using PhosphorImager analysis. Coomassie staining was used to demonstrate equal protein loading. Kinase assays were performed in duplicate using whole cell extracts from two independent experiments.
Results
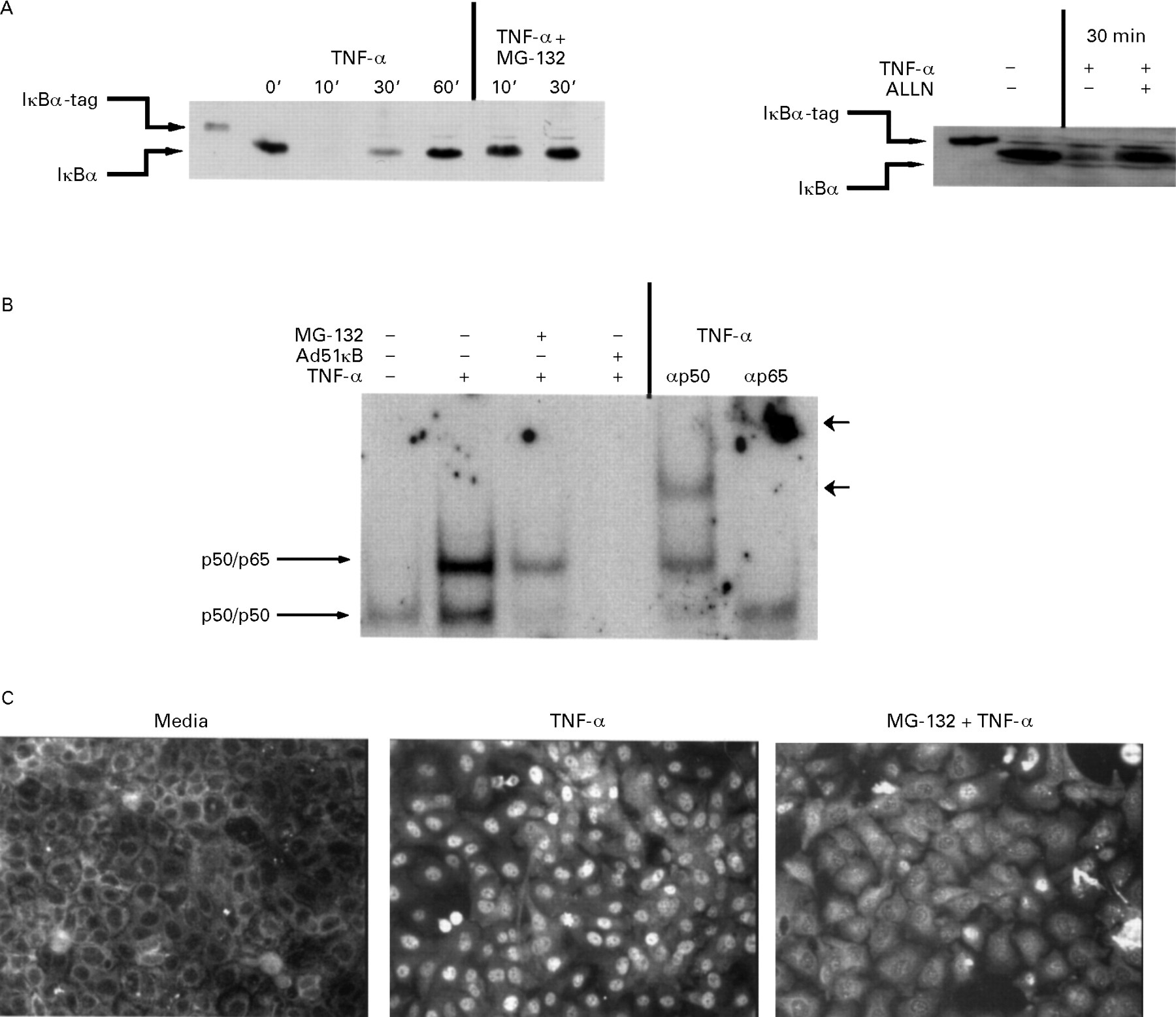
We investigated the regulation of ICAM-1 gene expression in TNF-α stimulated IEC-6 cells by studying activation of the IκB/NF-κB system and the consequences of blocking this system. We first analysed cytoplasmic concentrations of the NF-κB inhibitor, IκBα protein, at various time points after TNF-α stimulation. IEC-6 cells were stimulated with an optimal dose of TNF-α (10 ng/ml) for up to 60 minutes, and steady state concentrations of IκB protein were measured at various time points by western blotting. Immunoreactive IκBα rapidly disappeared within 10 minutes of TNF-α treatment and reappeared by 60 minutes (fig 1A). Pretreatment of IEC-6 cells with the protein inhibitor cycloheximide completely prevented the reappearance of IκBα at 60 minutes, indicating that IκB replenishment was due to new protein synthesis (data not shown). IκBα degradation in TNF-α stimulated IEC-6 cells was blocked by pretreatment with MG-132 or ALLN (fig 1A), which inhibit proteolytic cleavage of IκBα without interfering with phosphorylation.27 ,32 ,38-42

Effect of tumour necrosis factor α (TNF-α) stimulation and selective proteasome blockade on steady state IκBα concentrations (A), NF-κB binding activity (B), and RelA (p65) subcellular localisation (C). (A) Cells were pretreated with MG-132 (5 μg/ml; left panel), ALLN (50 μg/ml; right panel), or medium alone for 30 minutes and then stimulated for 0 to 60 minutes with TNF-α (10 ng/ml). Total protein was extracted and 20 μg subjected to SDS/PAGE followed by immunoblotting of IκBα using the ECL technique as described in the Methods section. Note that the IκBα standard used as a control contains seven extra amino acids (IκB-tag) compared with the endogenous epithelial IκBα. (B) Cells were treated as described above or infected for 12 hours with the Ad5IκB virus and then stimulated for 30 minutes with TNF-α (10 ng/ml). Nuclear extracts (5 μg) were tested for κB binding activity by electrophoretic mobility shift assay. Antibody supershifting is indicated by arrowheads. (C) Cells were treated as described in (A) and RelA localisation was visualised using an anti-RelA antibody followed by a rhodamine conjugated detection antibody.
Degradation of IκBα in TNF-α stimulated IEC-6 cells suggests that NF-κB could be translocated into the nucleus. We determined the presence of NF-κB in the nucleus in TNF-α stimulated cells with or without MG-132 pretreatment by EMSA and immunofluorescence staining. Nuclear extracts from TNF-α stimulated IEC-6 cells showed strong DNA binding activity compared with nuclear extracts derived from unstimulated cells (fig 1B). Antibody supershifting analysis shows that both the p50/p65 heterodimer and the p50/p50 homodimer were induced in TNF-α stimulated IEC-6 cells (fig 1B). Nuclear NF-κB activity was strongly reduced by MG-132 treatment (fig 1B) and to a lesser extent by ALLN (data not shown). Expression of the NF-κB super-repressor (Ad5IκB), which is resistant to phosphorylation and subsequent degradation because of serine to alanine mutations at positions 32 and 36,28 completely blocked TNF-α stimulated NF-κB nuclear complex binding activity (fig 1B). Moreover, immunofluorescence staining using the RelA antibody showed nuclear staining in the TNF-α treated cells compared with cytoplasmic staining seen in cells from untreated medium (fig 1C). Nuclear translocation of RelA was considerably attenuated by MG-132 pretreatment (fig 1C). Similar results were obtained with ALLN (data not shown). These results show that TNF-α rapidly activates NF-κB in IEC-6 cells by inducing IκBα degradation, and that blockade of IκBα degradation by several proteasome inhibitors prevents NF-κB activity.
We then investigated whether proteasome inhibitors act by selectively inhibiting IκBα degradation or by interfering with more proximal TNF-α signalling pathways. Membrane receptor bound TNF-α signals are transduced through multiple second messengers including the JNK/SAPK pathway.43-45 Activation of JNK/SAPK leads to the phosphorylation and activation of c-Jun.46 ,47 To test the specificity of NF-κB inhibition by ALLN, we investigated the activity of JNK/SAPK in IEC-6 cells using GST-c-Jun as a substrate as described previously.44 ,46 Cell extracts prepared from TNF-α stimulated IEC-6 cells showed a two- to three-fold stimulation of JNK/SAPK activity (fig 2; compare lanes 1 and 3), which was not inhibited by ALLN (fig 2; compare lanes 3 and 4). Similar results were obtained using a rat hepatic stellate primary cell culture system.48 Whole cells extracts incubated with the mutated substrate (GST-c-JunAA) or GST alone had no activity (data not shown). These results show that ALLN does not globally inhibit TNF-α signalling pathways but specifically inhibits the IκB/NF-κB system.

ALLN does not interfere with tumour necrosis factor α (TNF-α) induced JNK/SAPK activity in IEC-6 cells. Cells were pretreated with ALLN (50 μg/ml) or medium alone for 30 minutes and then stimulated with TNF-α (10 ng/ml) for 30 minutes. Phosphorylated glutathione S-transferase (GST)-c-Jun was visualised after protein fractionation using SDS/PAGE (12.5% gel) and quantified using PhosphorImager analysis. Coomassie staining was used to confirm equal protein loading. A representative result of three independent experiments is shown.
The ICAM-1 gene promoter has potential binding sites for NF-κB. Therefore we examined the effects of TNF-α stimulation and proteasome blockade on ICAM-1 expression in IEC-6 cells. Cells were pretreated with ALLN or vehicle for 30 minutes and then stimulated for four hours with TNF-α, after which ICAM-1 mRNA expression was evaluated by RT-PCR. As shown in fig 3A (upper panel), ICAM-1 mRNA was strongly up regulated after TNF-α incubation (compare lanes 1 and 2) and was almost completely inhibited by ALLN treatment (compare lanes 2 and 3). The other proteasome inhibitor MG-132 also caused a down regulation of ICAM-1 gene expression (fig 3A lower panel). These changes in ICAM-1 mRNA accumulation were not due to unequal RNA loading since the amount of co-amplified actin mRNA showed little variation (fig 3A).

Inhibition of ICAM-1 gene expression by proteasome inhibitors in IEC-6 cells as measured by RT-PCR (A) and ELISA (B) techniques. (A) Cells were pretreated with ALLN (50 μg/ml; upper panel), MG-132 (5 μg/ml; lower panel), or medium alone for 30 minutes and then stimulated with tumour necrosis factor α (TNF-α) (10 ng/ml) or medium alone for four hours. Total RNA was extracted, reverse transcribed, and amplified using specific ICAM-1 or actin primers. PCR products were run on a 2% agarose gel and stained with ethidium bromide. These results are representative of three different experiments. (B) Cells were stimulated with TNF-α (10 ng/ml) or medium alone for 16 hours in the presence or absence of ALLN (50 μg/ml) or aprotinin (10 μg/ml). Immunoreactive ICAM-1 protein synthesis was measured as described in the Methods section. Data represent mean (SE) from four samples. *p<0.05 compared with control samples. This result is representative of three different experiments.
ICAM-1 expression was also evaluated at the protein level. IEC-6 cells were stimulated with TNF-α (10 ng/ml) for 16 hours in the presence or absence of ALLN (50 μg/ml), and ICAM-1 protein levels were measured by ELISA. Figure 3B shows that TNF-α treatment of IEC-6 cells resulted in a significant induction of ICAM-1 protein synthesis. This up regulation of ICAM-1 protein production was completely inhibited in ALLN pretreated cells, consistent with our findings at the mRNA level (fig 3A). The serine protease inhibitor aprotinin failed to prevent TNF-α stimulation of ICAM-1 protein (fig 3B) and IκBα degradation (data not shown), demonstrating the specificity of ALLN on ICAM-1 gene expression. We further confirmed TNF-α stimulated ICAM-1 protein synthesis in IEC-6 cells by immunohistochemical analysis. TNF-α treated cells (fig 4A) showed increased ICAM-1 staining compared with unstimulated cells (fig 4B). No immunoreactivity was observed when non-specific mouse IgG was used (data not shown). These results show that TNF-α induces ICAM-1 gene expression at the mRNA and protein level, a process that is inhibited by proteasome inhibitor treatment.
Tumour necrosis factor α (TNF-α) induces ICAM-1 protein systhesis in IEC-6 cells as measured by immunohistochemical analysis. Cells were grown on glass coverslips and then stimulated with TNF-α (10 ng/ml) (B) or cultured in medium alone (A) for 16 hours. The cells were stained for ICAM-1 using anti-ICAM-1 antibody as described in the Methods section. Similar results were obtained in two other experiments.
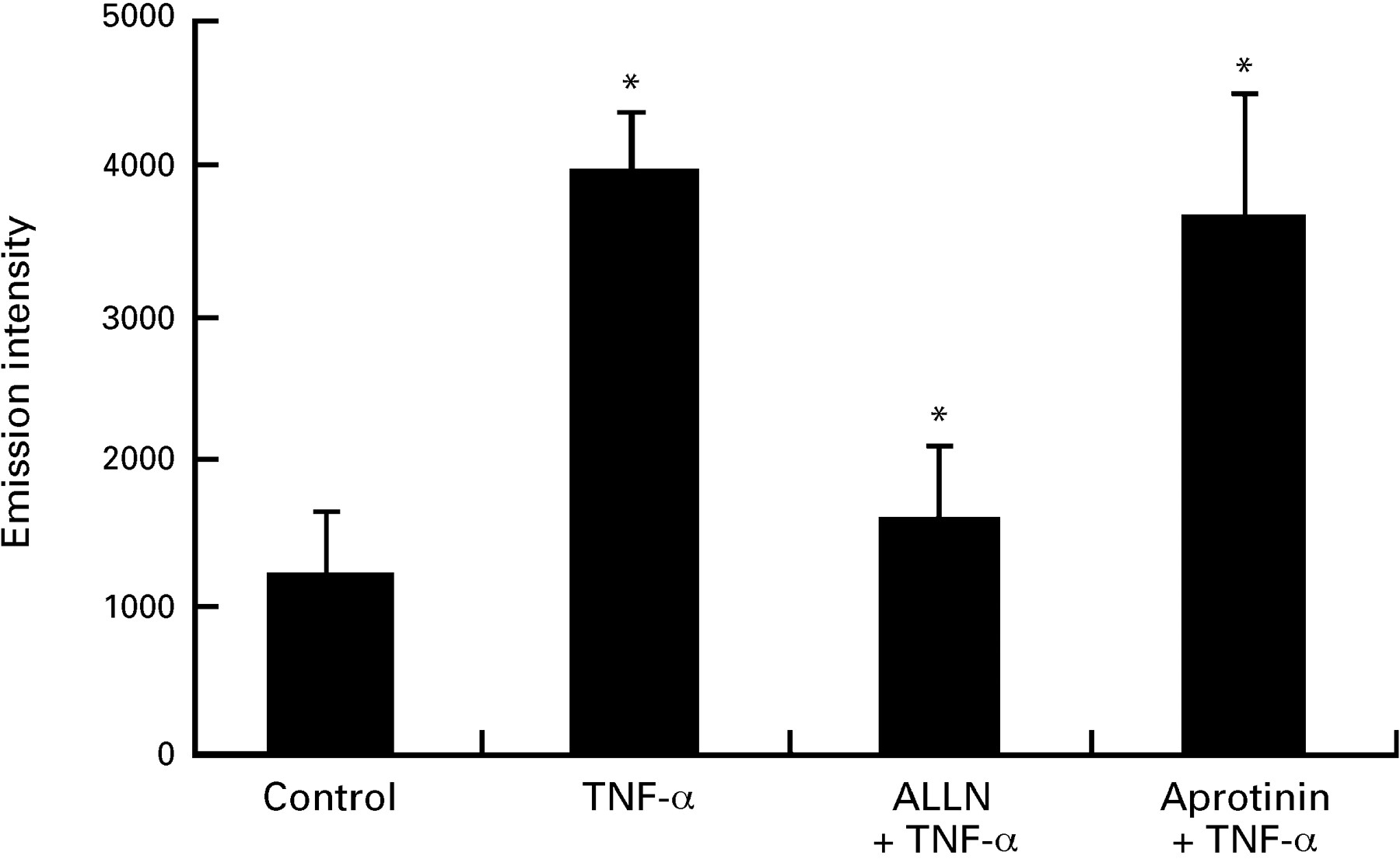
A possibility exists that the proteasome inhibitors non-specifically down regulate new protein synthesis and therefore prevent ICAM-1 gene induction by an NF-κB independent mechanism. To test this, IEC-6 cells pretreated with medium or MG-132 were labelled with [35S]methionine for 12 hours in the presence or absence of TNF-α (10 ng/ml). As a positive control, we used the protein synthesis inhibitor cycloheximide (50 μg/ml). As shown in fig 5A, MG-132 does not inhibit de novo protein synthesis in IEC-6 cells as opposed to the almost complete inhibition in cells treated with cycloheximide. In addition, no sign of protein breakdown after MG-132 treatment was noticed on a Coomassie blue stained gel (fig 5B). These data indicate that MG-132 does not interfere with the basic protein synthesis machinery. Since ICAM-1 is a ligand involved in adhesion of T cells and neutrophils, we next investigated whether the ICAM-1 up regulation found in IEC-6 cells would result in increased binding of a lymphocyte cell line (MOLT-4) and whether ALLN treatment would modulate this binding. As shown in fig 6, the binding of MOLT-4 cells to TNF-α stimulated IEC-6 cells was increased 3-fold compared with unstimulated cells. Pretreatment of IEC-6 cells with ALLN notably inhibited this TNF-α induced MOLT-4 adhesion to IEC-6 cells. The non-selective serine protease inhibitor aprotinin failed to block TNF-α induced MOLT-4 adhesion, showing the specificity of ALLN.

Effect of MG-132 on IEC-6 cellular protein synthesis. Cells were starved for 60 minutes in medium without methionine and then labelled for 12 hours with 150 μCi [35S]methionine in the presence or absence of MG-132 or cycloheximide (CHX). Total protein was extracted and 20 μg subjected to SDS/PAGE. (A) Gel exposed to autoradiography or (B) stained with Coomassie blue as described in the Methods section. Similar results were observed in two other independent experiments.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ALLN blocks the tumour necrosis factor α (TNF-α) mediated adhesion of IEC-6 to MOLT-4 lymphocytes. Fluorescence labelled MOLT-4 cells were added to TNF-α stimulated IEC-6 cells pretreated with ALLN (50 μg/ml), aprotinin (10 μg/ml), or medium alone. Fluorescence emission intensity in cell lysates after washing was measured as described in the Methods section. Similar inhibition was observed in two other independent experiments.
Discussion
ICAM-1 mediates adhesion of leucocytes to endothelial and epithelial cells during their migration to inflammatory foci and may be involved in the elaboration and persistence of inflammation in various diseases. ICAM-1 expression has been investigated in many cell systems including human transformed IECs, but the regulation of ICAM-1 expression in IECs has not been previously determined. In this study, we show for the first time that ICAM-1 gene expression is induced by TNF-α stimulation in rat non-transformed IEC-6 cells. In addition, we show that TNF-α activates NF-κB by inducing rapid IκBα degradation in IEC-6 cells, and provided direct evidence that NF-κB plays a crucial role in TNF-α induction of ICAM-1 gene expression on the basis of inhibition of ICAM-1 expression with pharmacological blockade of IκBα degradation using two independent proteasome inhibitors. These results are in agreement with the ability of proteasome inhibitors to block ICAM-1 expression in cultured endothelial cells.33 ,34
ICAM-1 mRNA accumulation is strongly up regulated in IEC-6 cells after TNF-α stimulation. This mRNA accumulation is translated into protein since we observed a twofold induction of ICAM-1 synthesis as determined by ELISA. This induction is similar to that seen in human HT-29 cells.3 ,4 The rapid ICAM-1 gene up regulation (four hours) argues against an autocrine mechanism involving other cytokines, especially IL-6, since the kinetics of ICAM-1 and IL-6 mRNA expression are similar in TNF-α stimulated IEC-6 cells.49 ,50Confirmation of enhanced ICAM-1 protein synthesis and function in response to TNF-α stimulation is provided by immunohistochemical staining and cellular adherence. Adhesion of the lymphocyte cell line MOLT-4 to IEC-6 cells is strongly induced by TNF-α stimulation, with functional cell adhesion increased to a greater degree than the induction of ICAM-1 protein. This may suggest the involvement of other adhesion molecules such as ICAM-2, ICAM-3, or vascular cell adhesion molecule-1 (VCAM-1) in the mediation of epithelial cell-immune cell adhesion after exposure to proinflammatory cytokines.
Our study showed an early and complete degradation of IκBα in TNF-α stimulated IEC-6 cells. These results are in contrast with our previous observation that most human transformed and native colonic epithelial cells, with the notable exception of Caco-2 cells, have delayed incomplete IκBα degradation in response to IL-1β and TNF-α, despite NF-κB nuclear translocation and inhibition of IL-8 secretion by blockade of IκBα degradation.27 Thus IEC-6 and Caco-2 cells have similar IκBα degradation responses, which are more like haematopoietic cells than most IECs.
Modulation of proinflammatory molecule synthesis is a key strategy in down regulating the inflammatory process found in various diseases such as inflammatory bowel disease (IBD). Synthesis of many cytokines and adhesion molecules is controlled at the level of transcription by NF-κB.14 ,16 ,17 Our strategy was to inhibit NF-κB activity and consequently decrease proinflammatoy gene expression by blocking IκBα degradation. This approach is conceptually superior to blocking individual inflammatory mediators because it targets a key transcriptional regulator of multiple proinflammatory genes and therefore interrupts the downstream inflammatory cascade. To achieve this goal, we focused our attention on the proteasome complex, which is involved in IκBα degradation.23-25 ALLN and MG-132 exert a strong inhibitory effect on proteasome activity and thereby inhibit NF-κB activity, although MG-132 is a more specific proteasome inhibitor than ALLN, with inhibition achieved at doses as low as 10 μM.23 ,24 ,29-32 Importantly, proteasome inhibitors do not interfere with the upstream signal leading to IκBα phosphorylation.38 ,39 We showed that a short preincubation (30 minutes) with two proteasome inhibitors with different specificities was able to block TNF-α induced IκBα degradation in IEC-6 cells almost completely. The rapid inhibition of IκBα degradation (10 minutes) suggests that ALLN and MG-132 act on a pre-existing protease involved in IκB degradation, probably the chymotrypsin-like activity. The consequences of IκB degradation blockade were the reduction of NF-κB activation, as observed by both EMSA and immunofluorescence, and the almost complete inhibition of ICAM-1 mRNA expression, protein synthesis, and lymphocyte binding activity. When ICAM-1 expression was blocked by ALLN, adhesion of MOLT-4 to IEC-6 cells was almost totally abolished.
Expression of adhesion molecules in patients with active IBD has been reported.51 The potential of IECs to produce ICAM-1 may have an impact on the onset of intestinal inflammation. For example, interaction of IECs with leucocytes has been shown to modulate the state of activation of IECs.52 ,53 In addition, an ICAM-1 antisense oligonucleotide was effective in preventing and reversing colitis in an animal model.54 The relevance of ICAM-1 in human IBD is shown by the preliminary report of the efficacy of an ICAM-1 antisense oligonucleotide in treating a small number of steroid dependent or refractory patients with Crohn’s disease.13Preliminary studies indicate activation of NF-κB in active Crohn’s disease, ulcerative colitis, and inflammatory control compared with inactive IBD and normal controls.55 ,56 We have previously shown that blockade of IκBα degradation strongly inhibits cytokine induced IL-8 secretion by IECs,27 thereby eliminating a key chemotactic signal for leucocyte migration.57 These in vitro protective effects are consistent with the preliminary report of dramatic attenuation of experimental granulomatous colitis by in vivo proteasome blockade.58 Therefore broadly blocking expression of a number of adhesion molecules and proinflammatory genes with a proteasome inhibitor can have extremely potent therapeutic effects.
Inhibition of the potent p65 transactivator was demontrated by EMSA and indirect immunofluorescence. Interestingly, RelA (p65) is the major NF-κB subunit mediating induction of ICAM-1 gene transcription in endothelial cells.15 Therefore we believe that the almost complete inhibition of nuclear RelA translocation and RelA DNA binding activity may explain the total blockade of ICAM-1 gene expression. This concept is supported by the dramatic in vivo effects of p65 (RelA) antisense oligonucleotide therapy in experimental colitis.59
The specificity of ALLN and MG-132 for the IκB/NF-κB system was evaluated at several levels. First, we investigated the effect of ALLN on an early event in the TNF-α signal transduction cascade by measuring the activation of JNK/SAPK. The phosphorylation of c-Jun has been shown to be an essential step in activation of this transcription factor.46 ,47 Increased phosphorylation of a recombinant GST-c-Jun construct in TNF-α stimulated IEC-6 cells indicates increased JNK/SAPK activity. This TNF-α induced kinase activity was not inhibited by ALLN treatment. Secondly, we looked at the ability of the serine protease inhibitor aprotinin, which does not possess any anti-proteasome bioactivity, to block ICAM-1 gene expression. Aprotinin failed to inhibit TNF-α induction of ICAM-1 protein, as measured by ELISA, and did not prevent the induction of lymphocyte adhesion to IEC-6 cells. Thirdly, a highly selective proteasome inhibitor, MG-132, had identical activity. Fourthly, total protein synthesis was not inhibited by MG-132. These results argue against a non-specific general inhibitory effect of ALLN and MG-132 on the TNF-α signalling pathway in IEC-6 cells.
In summary, our results show that TNF-α up regulates ICAM-1 gene expression in IEC-6 cells. NF-κB plays a major role in the ICAM-1 induction, as shown by inhibition of ICAM-1 gene expression and function when IκB degradation is selectively blocked. We believe that inhibition of a number of proinflammatory molecules by targeting the IκB/NF-κB system represents an exciting and promising approach to the treatment of intestinal inflammatory disorders.
Acknowledgments
This work was supported by NIH grants DK 47700, DK 34987, AI 26774, and GM 41804. C H was supported by a grant from the Deutsche Forschungsgemeinshaft (He 2458/1–1).