Article Text

Abstract
NF-κB is a pleiotropic transcription factor with key functions in the intestinal immune system. NF-κB family members control transcriptional activity of various promoters of proinflammatory cytokines, cell surface receptors, transcription factors, and adhesion molecules that are involved in intestinal inflammation. The perpetuated activation of NF-κB in patients with active inflammatory bowel disease suggests that regulation of NF-κB activity is a very attractive target for therapeutic intervention. Such strategies include antioxidants, proteasome inhibitors, inhibition of NF-κB by adenoviral IκBα expression vectors, and antisense DNA targeting of NF-κB. These approaches will hopefully permit the design of new treatment strategies for chronic intestinal inflammation.
Statistics from Altmetric.com
Much recent research has focused on the regulation of cytokine gene expression by transcription factors in the mucosal immune system. In this review we will discuss the role of the transcription factor nuclear factor κB (NF-κB) in immune and inflammatory responses in the gut.
Members of the NF-κB family of transcription factors
Nuclear factor κB (NF-κB) designates a group of transcription factors defined in part by their ability to bind a specific DNA sequence first identified in the enhancer of the immunoglobulin κ light chain gene.1-6 In mammals the NF-κB family consists of several proteins including NF-κB1 (p50; precursor protein: p105), NF-κB2 (p52; precursor protein: p100), p65 (RelA), c-Rel (Rel), and RelB which share the so-called Rel homology domain. Furthermore, Rel proteins such as dorsal, Dif and Relish have been identified in Drosophila. The roughly 300-amino-acid N-terminal Rel homology domain of NF-κB family members mediates DNA binding, dimerisation, and interaction with an inhibitor protein called IκB.7 ,8
NF-κB can be found in the cytoplasm of most cells as an inactive complex with unprocessed precursor proteins (e.g. p105) or IκB (e.g. IκBα) proteins.1-6 Activation of cells with various stimuli then initiates a signalling cascade that finally leads to the disruption of the inactive complex and the release of NF-κB (fig 1).9-11 In lymphocytes, NF-κB can be released by stimulating cells with various agents such as lipopolysaccharide (LPS), phorbol 12-myristate 13-acetate (PMA), phytohaemaglutinin (PHA), immunoglobulin receptor-crosslinking, interleukin 2, and crosslinking of surface CD3 or CD28. Upon activation, NF-κB translocates into the nucleus and binds to DNA. The prototypical NF-κB is a heterodimer composed of the p50 and p65 subunits and the latter is the most frequent component of active NF-κB in humans. p65 containing complexes bind with high affinity to the consensus DNA sequences 5′-GGGPuNNPyPyCC-3′ (p65/p50) or 5′-GGGPuNPyPyCC-3′ (p65/c-Rel) leading to activation of transcription.12 ,13 In addition to the p50/p65 heterodimer, many other heterodimers or homodimers (e.g. p50) of NF-κB/Rel family members have been described. Interestingly, homodimers of the p50 subunit are constitutively present in nuclear extracts of lymphocytes. Their function, however, remains to be determined as p50 lacks a transactivation domain.
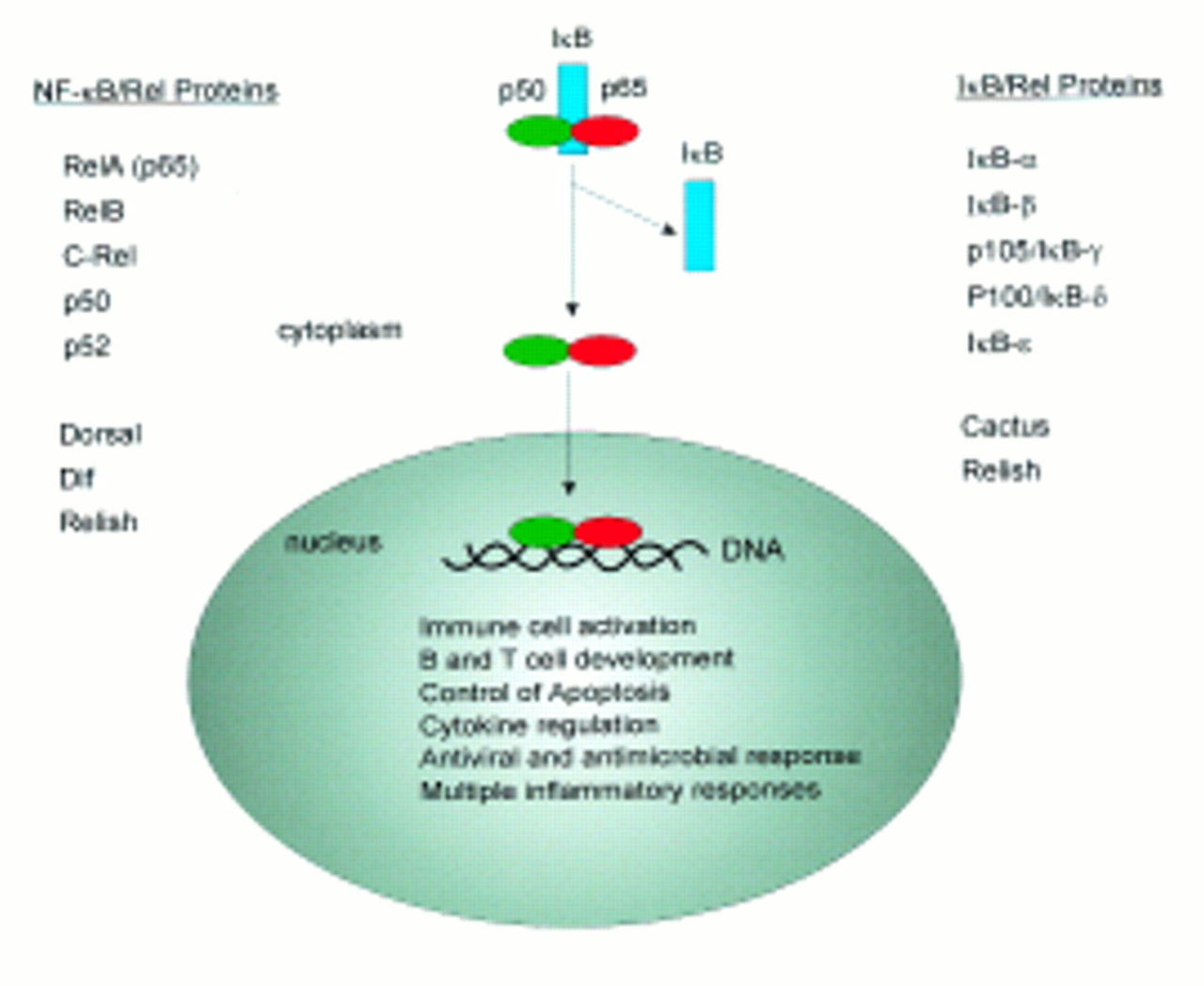
Members of the NF-κB/IκB families. After activation of the cells IκB is degraded and NF-κB can translocate to the nucleus.
The IκB family
The IκB family of proteins includes IκBα (MAD-3, pp40), IκB-β, IκBγ/p105, Bcl-3, IκBδ/p100, and IκBε.14-17 These proteins are characterised by multiple5-7 repeated sequences of 33 amino acids, termed SWI6/ankyrin repeats, which seem to be responsible for the interaction with the Rel domain of NF-κB. IκB proteins are organised as tripartite molecules consisting of (i) an N-terminal domain required for proteolytic degradation, (ii) a central domain with ankyrin repeats required for interaction with NF-κB, and (iii) a C-terminal domain (called PEST domain) which is essential for sequestration of NF-κB in the cytoplasm. The precursor proteins of p50 and p52, termed p105/IκBγ and p100/IκBδ, contain in addition to the Rel homology domain ankyrin repeats and thus are structurally and functionally related to the IκB family. For instance, the precursor of p50 (p105) contains at its N-terminal domain p50 and in its C-terminal half seven ankyrin repeats.18-20 This protein sequesters p65, c-Rel and proteolytically released p50. Although the p50 releasing protease of p105 has not yet been identified, an ATP dependent ubiquitin system for p105 has been proposed.
IκB proteins exert multiple functions including prevention of nuclear translocation of NF-κB. The inhibition is based on the interaction between the C-terminal ankyrin repeats of IκB and the Rel homology domain of NF-κB. The IκBα and IκBβ proteins preferentially inhibit NF-κB complexes containing the p65 and c-Rel subunits. Interestingly, some IκB proteins have been found in the nucleus. This fact suggests that these proteins do not necessarily reside as an anchor in the cytoplasm to fulfil their function. In fact, the IκB-like protein Bcl-3 can function as transcriptional coactivator after association with p52 on the DNA although Bcl-3 has been shown to inhibit p50 containing complexes.21 ,22 Furthermore, it has been proposed that IκB proteins are able to strip off DNA bound NF-κB.
Transcriptional regulation of NF-κB and IκB genes
p105 and p100 are constitutively expressed but their mRNA levels are increased in response to signals activating NF-κB, such as treatment with PMA, interleukin 1 (IL-1), and tumour necrosis factor (TNF). There is some evidence for autoregulation of NF-κB activity as the promoters of p105, c-Rel and Bcl-3 contain cis-acting κB motifs.23 ,24 In contrast, the transactivating p65 subunit is not induced by NF-κB and the p65 promoter does not contain NF-κB binding sites.25 However, NF-κB activation relies primarily on a rapid post-translational mechanism. Finally, it should be mentioned that NF-κB upregulates transcription of its inhibitor IκBα most likely due to its binding to κB sites at the IκBα promoter.26
Signal transduction pathways leading to NF-κB activation
Several unrelated stimuli like phorbol ester, TNF, IL-1, IL-18, LPS, and ultraviolet light have been shown to activate NF-κB, which is in agreement with the pleiotropic roles of NF-κB in many different cell types and tissues. Activation of NF-κB by IL-1, TNF and IL-18 requires binding of cytokines to their specific cell surface receptors (fig 2). For instance, TNF binds to its receptor and leads to activation of TNF receptor associated factor (TRAF) proteins via a receptor associated adaptor protein called TNF receptor associated death domaine (TRADD). At the moment six members of the TRAF protein family are known (TRAF 1–6). TRAF2 is required for NF-κB activation via TNFR1 (75 kDa) and TNFR2 (55 kDa). In contrast, TRAF5 is also involved in NF-κB activation by other members of the TNF receptor family and TRAF6 participates in NF-κB activation via IL-1.27 Furthermore, TRAF proteins interact directly with the cytoplasmic tails of two other TNFR family members (CD40 and CD30).28 ,29 TNFR1 mediated NF-κB activation also requires the serine-threonine kinase RIP, which is associated with TRAF proteins and interacts with the respective receptor complex via TRADD.30 TNF signalling also activates JNK (stress activated protein kinase) and Fas associated protein with a death domaine (FADD) which leads to apoptosis via a caspase-8 initiated cascade (fig 2).31-33

NF-κB signal transduction pathways initiated by IL-1, TNF and IL-18. Whereas TNF activates NIK via TRADD and RIP/TRAF2, IL-1 and IL-18 use IRAK/TRAF6 to activate NIK. NIK in turn activates IKKα which causes phosphorylation of IκB. Next, IκB is ubiquitinated and degraded via the proteasome pathway. Finally, NF-κB translocates into the nucleus and binds to its target DNA sequences.
Members of the TNF receptor (TNFR) superfamily interact via their cytoplasmic tails with TRAF proteins which serve as adaptor proteins to recruit NIK, a specific NF-κB inducing kinase.34 In addition to the TNF signalling pathway, the IL-1 and IL-18 initiated signalling pathways lead to the activation of NIK. However, in the case of IL-1 NIK is activated through TRAF6 and IRAK (serine-threonine kinase) and a similar activation mechanism of NF-κB has been recently suggested for IL-18 (fig 2). NIK is classified as MAP kinase kinase kinase (MAP3k) and was identified as TRAF2 interacting protein.35 A serine-threonine kinase previously known as CHUK was shown to associate with NIK and IκBα in mammalian cells.36 Based on its property to phosphorylate IκBα CHUK was named IKKα (IκB kinase α).37 At the next step of the cascade IKKα associates with IκBα and phosphorylates the latter at serine 32 and serine 36. The modified IκBα is then specifically degraded via the ubiquitin/proteasome pathway and the active NF-κB dimer can translocate into the nucleus and bind to its cognate target sequence (fig3).38 ,39

{kind=link}
{kind=link}
{kind=link}
Targeting of the NF-κB activation pathway in intestinal inflammation. While alkylating agents and antioxidants may block protein kinases, antisense DNA can inhibit translation of p65. In addition, corticosteroids lead to blockade of p65 and adenoviral expression vectors could deliver genes whose products inactivate NF-κB.
Genes regulated by NF-κB
NF-κB is a key regulator of the inducible expression of many genes associated with immune function in the gut. For instance, NF-κB plays an essential role in the transcriptional regulation of many cytokine genes (e.g. IL-1, interferon (IFN) γ, IL-2, IL-6, IL-8, IL-12p40) in lymphocytes, epithelial cells and monocytes. The role of NF-κB in IL-2 gene expression has been extensively studied in the past. Stimulation of T cells via the CD28 pathway leads to activation of NF-κB and subsequent binding to the CD28 response element (CD28RE) of the IL-2 promoter.40 Interestingly, such elements were also found upstream of the IL-3 gene and IL-8 expression is also regulated by NF-κB in response to CD28 co- stimulation.41 ,42 This suggests a common pathway of CD28 stimulated cytokine expression in T cells involving NF-κB. However, the potential role of NF-κB in immune modulation in the gut is not only limited to cytokine gene regulation. In fact, NF-κB has been shown to have an important function in the regulation of a variety of genes encoding transcription factors and cell adhesion molecules (table 1). For instance, the binding of NF-κB, activating transcription factor-2 (ATF-2) and high mobility group I(Y) (HMG-I(Y)) to the E-selectin promoter is necessary for the expression of the respective gene.43 ,44 Furthermore, NF-κB regulates the expression of genes for the transporter associated with antigen processing (TAP-1), the proteasome subunit latent membrane protein 1 (LMP-1) and the MHC class II invariant chain,45 ,46proteins with essential functions for antigen presentation. Thus, NF-κB seems to be a key regulator of immune cell function.
Genes regulated by NF-κB
NF-κB itself is extensively up- and downregulated by a wide variety of exogenous stimuli that modulate immune function, thus providing a positive or negative feedback mechanism. For instance, NF-κB transactivates the inducible nitric oxide (NO) synthase promoter in response to LPS giving rise to increased production of NO, a substance that is strongly upregulated in the inflamed intestine,47which in turn has been reported to inhibit NF-κB activation in endothelial cells.48 ,49 Interestingly, various other substances clinically used to treat patients with chronic intestinal inflammation, including IL-10, sulphasalazine and immunosuppressive drugs such as cyclosporin A and glucocorticoids, have been reported to inhibit NF-κB activation.50-55 ,74 Whereas corticosteroids repress NF-κB activity by inducing IκBα protein production and complex formation with NF-κB p65, the inhibitory mechanisms of IL-10 on NF-κB activation have not been fully delineated. However, IL-10 knockout mice with chronic intestinal inflammation have activated NF-κB p65.56 Furthermore, a phosphorothioate oligonucleotide antisense to the p65 translation start site suppresses colitis in these mice indicating an important role for IL-10 in controlling NF-κB activity in the gut.
Lessons from NF-κB/IκB gene knockouts
Recently, the targeted disruption of various genes encoding NF-κB subunits has been described. These knockout mice revealed severe defects in immune function further supporting a key regulatory role for NF-κB in the immune system. Interestingly, the phenotype of these mice differed strikingly depending on the disruption of the respective NF-κB subunit. For instance, mice lacking the p50 subunit (NF-κB1) developed normally but had severe defects in immune cell function.57 B cells of these mice had an impaired ability to produce antibodies and to proliferate upon LPS challenge. Furthermore, p50 −/− mice were highly susceptible to bacterial infections with staphylococcus and listeria. If compared with p50, the phenotype of p65 (RelA) knockout mice was even more dramatic. These animals died during embryonic development, most likely because of extensive apoptosis of cells in the liver.58 Analysis of NF-κB regulated genes (GM-CSF, IκBα) revealed a loss of inducibility in the p65 knockout mice and cultured T cells from these mice showed strikingly reduced proliferative responses, underlining the functional importance of NF-κB p65 for appropriate immune function.
Mice lacking RelB developed normally until days 8–10. Subsequently, however, they showed a complex pathological phenotype, which is the result of multiple defects in the adult immune system.59RelB −/− mice displayed T cell mediated inflammation of multiple organs and had impaired cellular immunity. Mice lacking c-Rel developed normally with no haemopoietic cell abnormalities.60Interestingly, mature T and B lymphocytes of c-Rel −/− mice had an impaired responsiveness to mitogenic stimuli like anti-CD3 or anti-IgM, respectively. Furthermore, in unchallenged animals immunoglobulin production was impaired. The proliferative block upon αCD3 and αCD28 stimulation of T cells disappeared after addition of exogenous IL-2, suggesting that c-Rel is necessary for high level IL-2 production.
In addition to knockout studies of NF-κB family members, several groups have focused their attention on the IκB family. IκB knockout mice displayed constitutively high nuclear levels of NF-κB, giving rise to a dramatic phenotype of these animals. Mice lacking IκBα, although apparently normal at birth, died approximately seven days later.61 Their phenotype showed small spleens and thymuses, skin defects and increased granulopoiesis. In addition, upregulated expression of some NF-κB regulated genes (G-CSF and VCAM-1, as defined in table 1) was observed. Taken together, the different phenotype of knockout mice of the NF-κB/IκB families indicates the unique function of each individual family member and shows that there is no simple redundancy among these proteins. Furthermore, the generation of NF-κB/IκB deficient mice has provided strong evidence for a key role of NF-κB in controlling multiple steps of immune cell function such as apoptosis, cytokine production and chronic inflammation.
Role of NF-κB in the mucosal immune system
Dysregulated cytokine production and signalling mechanisms by epithelial cells, mucosal lymphocytes and macrophages have been implicated in the pathogenesis of both Crohn’s disease and ulcerative colitis, the two major forms of human inflammatory bowel disease (IBD).62 Over the past few years, various murine models of chronic intestinal inflammation resembling IBD have been established. These models have provided important clues as to the nature of such dysregulation and to its possible cytokine based treatment.63 Thus, in studies of several of the models most closely resembling Crohn’s disease it was found that production of large amounts of Th1-type cytokines (e.g. IFNγ and TNF) whose promoters are regulated by NF-κB is a major and essential feature of the inflammation.64 ,65 Finally, it has been shown that Th1 cytokine production in these models is triggered by macrophages via increased production of IL-12, a cytokine that plays a major role in driving T cell differentiation and whose expression is also at least partially triggered by NF-κB.66
The above data encouraged studies on the identification of signalling pathways and transcription factors that govern cytokine gene transcription in IBD. Although some NF-κB family members are apparently important in preventing inflammatory responses (e.g. RelB), it was found that nuclear NF-κB levels are increased in patients with IBD.56 ,76 In particular, the p65 subunit was highly activated in epithelial cells and lamina propria macrophages from patients with active Crohn’s disease and ulcerative colitis.56 ,67 ,76 These findings are consistent with immunohistochemical data indicating increased expression of NF-κB p65 in active IBD68 ,76 and data from intestinal biopsy samples showing increased p65 in active Crohn’s disease.69 In addition, it was shown recently that a specific p65 antisense oligonucleotide can block p65 expression and proinflammatory cytokine production by lamina propria macrophages in patients with active Crohn’s disease and ulcerative colitis.56 Furthermore, in a murine model of colitis p65 antisense treatment led to an abrogation of chronic intestinal inflammation.56 In spite of these data on the role of NF-κB p65 in IBD, many additional questions have to be answered. In particular, there are few data concerning the role of other NF-κB/IκB family members in epithelial cells and T cells in the gut. In addition, the expression of IκB family members and their degradation mechanisms in IBD have only been partially characterised. Interestingly, recent data by Jobin and coworkers showed activation of NF-κB in epithelial cells in response to IL-1 and altered regulation of IκBα degradation in native colonic epithelial cells.67 Such enhanced resistance of epithelial cells to IκBα proteolysis suggested a potentially increased responsiveness to therapeutic blockade. Indeed, adenoviral mediated delivery of a mutant NF-κB repressing IκBα protein resulted in inhibition of IL-8 production by intestinal epithelial cells.70 Furthermore, pharmacological inhibition of IκBα degradation strongly reduced IL-8 secretion by intestinal epithelial cells.67 ,70 Finally, recent evidence suggests that NF-κB is important in regulating intercellular cell adhesion molecule (ICAM-1) expression in the intestine.71 ,75Preliminary data from the same group also showed a beneficial therapeutic effect of proteasome inhibitors (that block NF-κB activation) in experimental colitis.
Inhibition of NF-κB activity has been recently suggested as a major component of the anti-inflammatory activity of glucocorticoids that are frequently used for treatment of chronic intestinal inflammation in humans.72 ,73 Although activation of NF-κB p65 is not specific for patients with IBD, its perpetuated activation makes it a very attractive target for therapeutic intervention.74Thus, downregulation of NF-κB activity emerges as a potential key event in the control of chronic intestinal inflammation in humans and strategies to inhibit NF-κB activity more specifically are desirable. Such strategies include antioxidants, proteasome inhibitors, inhibition of NF-κB by adenoviral IκB expression vectors, and antisense DNA targeting of NF-κB p65 (fig 3). Thus, the above data suggest that targeting of NF-κB may be a novel molecular approach for the treatment of patients with IBD that could lead to the design of new treatment strategies that have added specificity but reduced toxicity compared with standard immunosuppressive therapy.