Article Text

Abstract
Background: Gliadins, a family of wheat proteins, are central to the pathogenesis of celiac disease (CD). In addition to ‘immunogenic’ effects, gliadin directly affects cultured cells and intestine preparations, and produces damage in vivo, via a separate ‘toxic’ peptide, such as A-gliadin p31–43 (P31–43).
Aims: Understanding the molecular mechanisms underlying direct non T-cell mediated effects of gliadin peptides, and assessing their potential role in promoting CD.
Method: Gliadin effects were tested on a number of cell lines and on cultured mucosa samples by evaluating cytoskeleton rearrangements, endocytosis, proliferation and apoptosis. Standard biochemical methods were used to assess prolonged epidermal growth factor receptor (EGFR) activation.
Results: Crude gliadin peptic-tryptic peptides (PTG], or P31–43 alone, fully reproduce the effects of epidermal growth factor (EGF] on actin cytosketon, cell cycle and cell proliferation of various cell lines. Inhibitor studies demonstrate the role of EGFR in the early response to gliadin exposure, pointing to activation of the EGFR pathway. Peptide P31–43 is not similar to any EGFR ligand, but can delay inactivation of the EGFR interfering with its endocytosis. Gliadin-induced delay of EGFR endocytosis in cultured intestinal biopsies, together with S-phase entry of epithelial intestinal cells, confirm a role for EGFR activation in CD.
Conclusion: The ability of gliadin peptides to delay EGFR inactivation through interference with the endocytic pathway suggests a model where gliadin fragments amplify the effects of trace amounts of EGF, and possibly of other growth factors, by prolonging receptor activation. The results, using cultures of coeliac intestinal biopsies, highlight the role of the EGF pathway in establishing and maintaining the typical atrophic and proliferative alterations of the small intestine in CD.
- CD, coeliac disease
- EGF, epidermal growth factor
- EGFR, epidermal growth factor receptor
- PDGF, Platelet derived Growth Factor
- PTG, peptic-tryptic digests of gliadin
- PTL, peptic-tryptic digest of lactoalbumin
Statistics from Altmetric.com
- CD, coeliac disease
- EGF, epidermal growth factor
- EGFR, epidermal growth factor receptor
- PDGF, Platelet derived Growth Factor
- PTG, peptic-tryptic digests of gliadin
- PTL, peptic-tryptic digest of lactoalbumin
Coeliac disease (CD), or gluten-sensitive enteropathy, is a multifactorial disease affecting approximately 1 out of 100 individuals in the European population and it is believed to be the result of a deregulated T-cell mucosal immune response to wheat gliadin and related prolamines of other cereals (barley and rye). This altered immune response has a strong genetic background with HLA genes playing a major role in susceptibility.1 Although there is strong evidence in favour of a skewed mucosal Th1 response to gliadin peptides,1 it is also likely that other non T-cell mediated phenomena play a role in the pathogenesis of the celiac lesion. Direct effects of gliadin peptides have been shown in several experimental systems: cultured cell lines,2–,6 foetal rat or chick intestine preparations and in vitro cultured biopsies from untreated CD patients.7,8,9,10 More recently, the ability of a peptic-tryptic digest of gliadin (PTG) to induce actin rearrangement has also been reported.11 Most of these studies highlight the function of an A-gliadin peptide, P31–43, from the N-terminal end of gliadin. Unlike peptide P56–68 which is one of the dominant epitopes recognised by T-cells isolated from the intestine of CD patients,12 P31–43 is not immunogenic for T-cells, but reproduces several effects of PTG, including the ability to agglutinate undifferentiated K562 cells, to induce and maintain tissue damage in small intestinal mucosa from patients with CD9,10,13 and to activate innate immune mechanisms in treated patients.14
In this paper a further investigation is made of the non-immunogenic activities of PTG and in particular of P31–43, such as induction of actin rearrangement and proliferative effects on cell lines and enterocytes. Our work has confirmed that gliadin peptides, and in particular P31–43, cause alterations of the cytoskeleton in different cell lines. In consideration of the fact that similar alterations have been described as caused by growth factors, this work verifies that the effects of gliadin peptides and P31–43, both on cell lines and cultured small intestinal biopsies, are mediated by epidermal growth factor receptor (EGFR) activation. In particular, this work shows that gliadin peptides interferes with EGFR endocytosis, amplifying the effects of epidermal growth factor (EGF). We hypothesise that these biological properties of gliadin peptides, and the mechanisms involved, are central to the pathogenesis of the coeliac lesion.
MATERIALS AND METHODS
Cell culture, treatments and cell staining
Caco-2 and NIH3T3 (CL7)15 cells were cultivated in Dulbecco Modified Eagle’s Medium DMEM, (GIBCO) 10% FCS (GIBCO), 100 units/ml penicillin-streptomycin (GIBCO), 1 mM glutamine.
LPS-free PT digests7 and synthetic peptides14 (Inbios, >95% purity, MALDI-toff analysis as expected) were obtained by Ultrasart-D20 (Sartorius AG, Goettingen, Germany) filtration. LPS levels were below detection (<0.20 EU/mg), assessed by commercial QCL-1000 kit (Cambrex Corporation, New Jersey USA). P31–43 sequence: LGQQQPFPPQQPY; P56–68 sequence: QLQPFPQPQLPY. Unless stated, peptic-tryptic digests of gliadin (PTG) and peptic tryptic digests of lactalbumin (PTL) were used at 500 μg/ml, P31–43 and P56–68 at 70 μg/ml, EGF at 100 ng/ml, EGF-Alexa488 at 20 ng/ml (Molecular Probes), ZD1839R (1 μM) and the blocking anti-EGFR528 mouse monoclonal antibody at a 1:100 dilution (clone 528, Santa Cruz). For actin staining,16 Caco-2 cells on glass cover slips kept for 1 h in low serum medium were treated with gliadin peptides for 15 min. and where required, EGFR inhibitors. Cover slips were fixed, permeabilised and stained with 0.1 mg/ml Texas Red–labelled phalloidin (Sigma Chemical Co.).16 Bromodeoxyuridine (BrdU) (Roche) incorporation and staining was performed as described elsewhere.15,16 Over 300 cells in several fields were evaluated in each sample. Variations were <25% of the mean. Images were generated with an Axiophot microscope (Carl Zeiss MicroImaging, Inc.) and processed with KS300 software (Carl Zeiss MicroImaging, Inc.). Confocal images were acquired with a LSM 510 Zeiss microscope.
Immunoprecipitation and immunoblotting
Cell lysates were prepared as described previously17,18 and the phosphorylated form of Erk [p-Erk] was detected using anti-p-Erk E-4 mouse monoclonal antibody (Clone E4 Santa Cruz). Total Erk was detected using Erk rabbit polyclonal antibody anti-Erk K23 (clone K23, Santa.Cruz). Lysates for EGFR endocytosis experiments were obtained and analysed as described elsewhere.19,20 Briefly, 2 micrograms of anti EGF-R immunoprecipitating mouse monoclonal antibody (clone 528, Santa Cruz) were added to 1 milligram of cell lysates in 500 micro litres of lysis buffer kept for 2 h at 4°C with gently rocking. Thirty micro litres of 1:1 protein-A agarose (Pierce) was added and the mixture was rocked gently for 1 h at 4°C, before centrifugation at 12 000 g for 5 min at 4°C. The immunoprecipitate was washed three times with 500 μl of lysis buffer. Proteins were separated by SDS-PAGE and immunoblotted with anti-phosphotyrosine (p-Tyr) mouse monoclonal antibody (clone 4G10 from UBI) or anti-EGFR rabbit polyclonal antibody (clone 1005, Santa Cruz). Electrophoresis and immunoblotting were all performed as described elsewhere.17 Densitometric analysis was performed as follows. Band intensity was evaluated by integrating all the pixels of the band without the background, calculated as the average of the pixels surrounding the band. Phosphorylation increments (Pi) were calculated from the ratio of phosphorylated (P) and unphosphorylated (UP) bands in treated (t) and untreated cells (ut) as follows: Pi = (Pt/UPt)/(Put/UPut).
Data bank analysis
Swissprot, Trembl and InterPro data banks were searched for sequences matching peptide P31–43, by using Blast and FastA. Sequence alignment was performed by using ClustalW and visualised by PrettyPlot from the EMBOSS suite.
Organ culture studies
All biopsies were treated as described previously.14,21 To examine endocytosis of EGF, six intestinal biopsies from untreated coeliac patients and six from control subjects affected by gastro-esophageal reflux were cultured for 3 hours with Alexa-488 fluorochrome labelled EGF (pulse) and then after careful washing, PTG or P31–43 or medium only was added. Three samples from untreated coeliac patients and three from controls were harvested after 6 h chase, three other samples from untreated CD patients and three from controls were harvested after 24 h chase. All samples were prepared for cryo-sectioning; air dried sections of 5 μm were analyzed with a confocal microscope.
For BrdU staining, duodenal biopsies from 10 untreated CD patients were cultured for 24 hours in the presence of BrdU, in medium alone (n = 10) or with PTG (n = 10) or P31–43 (n = 10)+/−anti-EGFR-528. As controls, four biopsies showing villous atrophy from untreated CD patients were cultivated with PTL, six with anti-EGFR-528 alone and three with P56–68; finally, from non-coeliac patients five biopsies were cultivated with PTG and three with P31–43 together with BrdU. Biopsies were than snap frozen and 5 μm air dried sections were stained for 1 hr with anti-cytokeratin mouse antibodies (Dako 1:100) followed by 1 h with secondary Texas red anti-mouse IgG (Jackson Immunoresearch) (2 μg/ml) at room temperature in a dark humid chamber to identify epithelial cells. BrdU staining was performed as described above. The number of BrdU positive cells divided by total cytokeratine positive cells resulted in the percentage of BrdU positive cells. Epithelial cell apoptosis was assessed by staining for cytokeratin, combined with the TUNEL-FITC assay (Roche).
Statistical analysis
Statistical analysis was performed where appropriate by employing Student’s t-Test; asterisks mark results where P<0.05.
RESULTS
EGFR activation mediates gliadin peptide-induced actin rearrangements
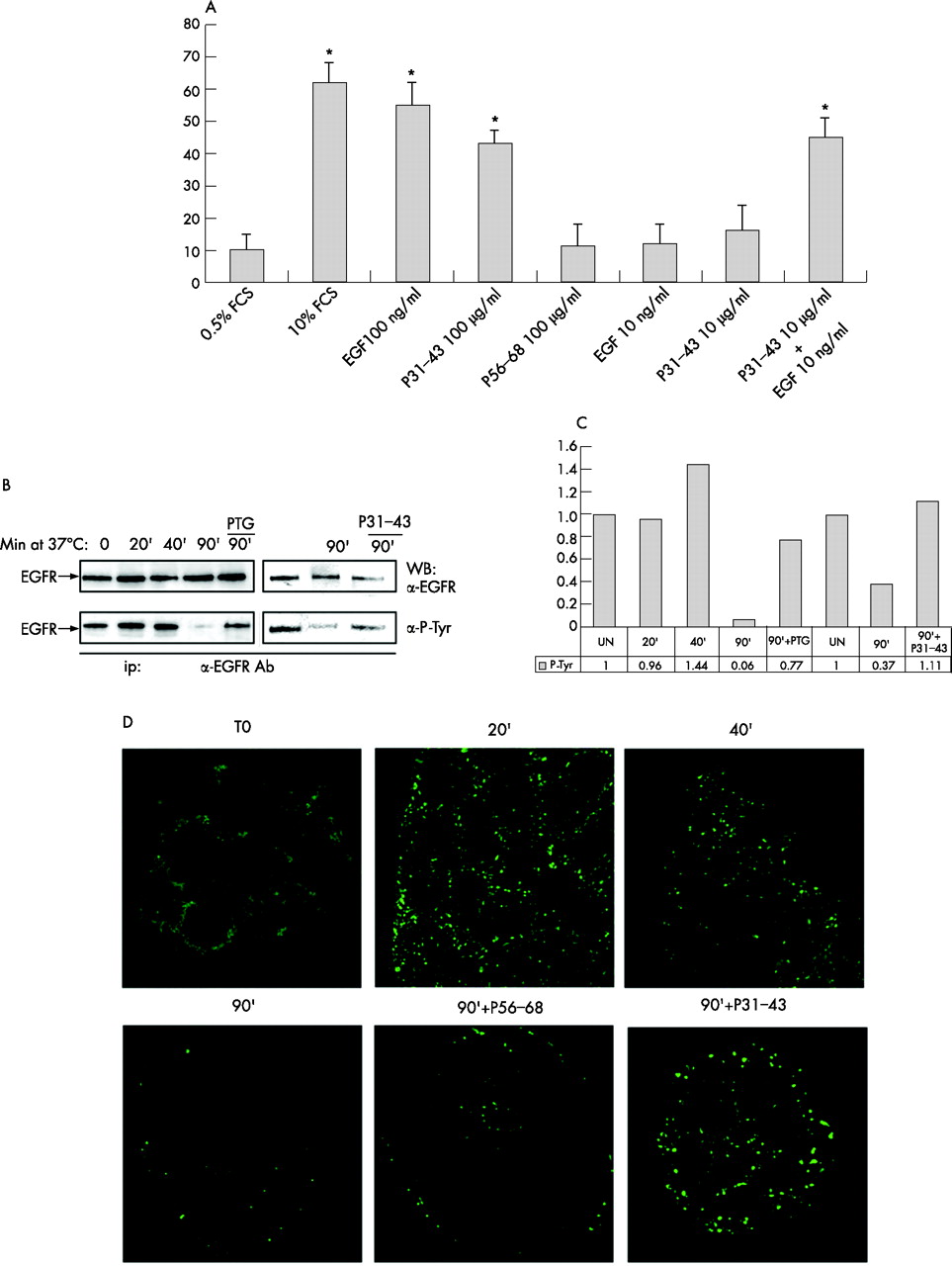
The effects of gliadin peptides on the actin cytoskeleton of Caco-2 cells, an epithelial cell line of intestinal origin, were tested. As shown in fig 1A⇓, B, 15 min. treatment with a PTG or with P31–43 induced large membrane rufflings at the edge of the clusters of Caco-2 cells. The effect is specific for PTG and P31–43, as it was not reproduced by a peptic-tryptic digest of lactalbumin or by P56–68, one of the immunogenic peptides for coeliac patients. These phenomena have already been described11 in intestinal cell lines and can be described in cell lines of different origin such as MCF7 cells derived from mammary carcinoma or fibroblasts NIH3T3 (not shown).

Peptic-tryptic digest of gliadin (PTG) and P31–43 effects on actin rearrangement depend on epidermal growth factor receptor (EGFR) activation. [A] Phalloidin stained Caco-2 cells 15 min. after addition of (PTG), peptic-tryptic digest of lactoalbumin (PTL), P31–43, P56–68 or epidermal growth factor (EGF). White arrows point to fan-like actin modifications appearing at the edge of Caco-2 clusters, after PTG, P31–43 or EGF treatment (100× objective). [B] Lower magnification with 40× objective of Caco2 clusters stained with phalloidin untreated and after 15′ of P31–43 treatment, large rufflings can be observed at the edge of the entire cluster. [C] Actin modification can be prevented by EGFR inhibitors. Representative results from 4 independent experiments.
Rapid and dramatic changes of the actin cytoskeleton are well-known effects of a number of growth factors, such as EGF and Platelet derived Growth Factor (PDGF), on several cell types.22 In support of this, EGF treatment produced actin rearrangements in Caco-2 cells very similar to those found with gliadin (fig 1A⇑). No similar effects were obtained with PDGF, insulin and Lisophosphatidic acid (LPA) (data not shown). To confirm that the effects of PTG and P31-43 involved the EGF signal transduction pathway, gliadin-stimulated Caco-2 cells were treated with two different EGF receptor (EGFR) inhibitors, ZD183923 and anti-EGFR-528.24 ZD1839 inhibits EGFR mediated tyrosine kinase activity by interacting with its autophosphorylation site; the anti-EGFR 528 antibody recognises the extra cellular ligand domain of EGFR and so prevents EGFR activation by acting as a ligand competitor. Both reagents completely blocked EGF induced actin rearrangements in Caco-2 cells (not shown), as well as those induced by PTG and P31-43, confirming that activation of the EGFR is needed in both processes (fig 1C⇑).
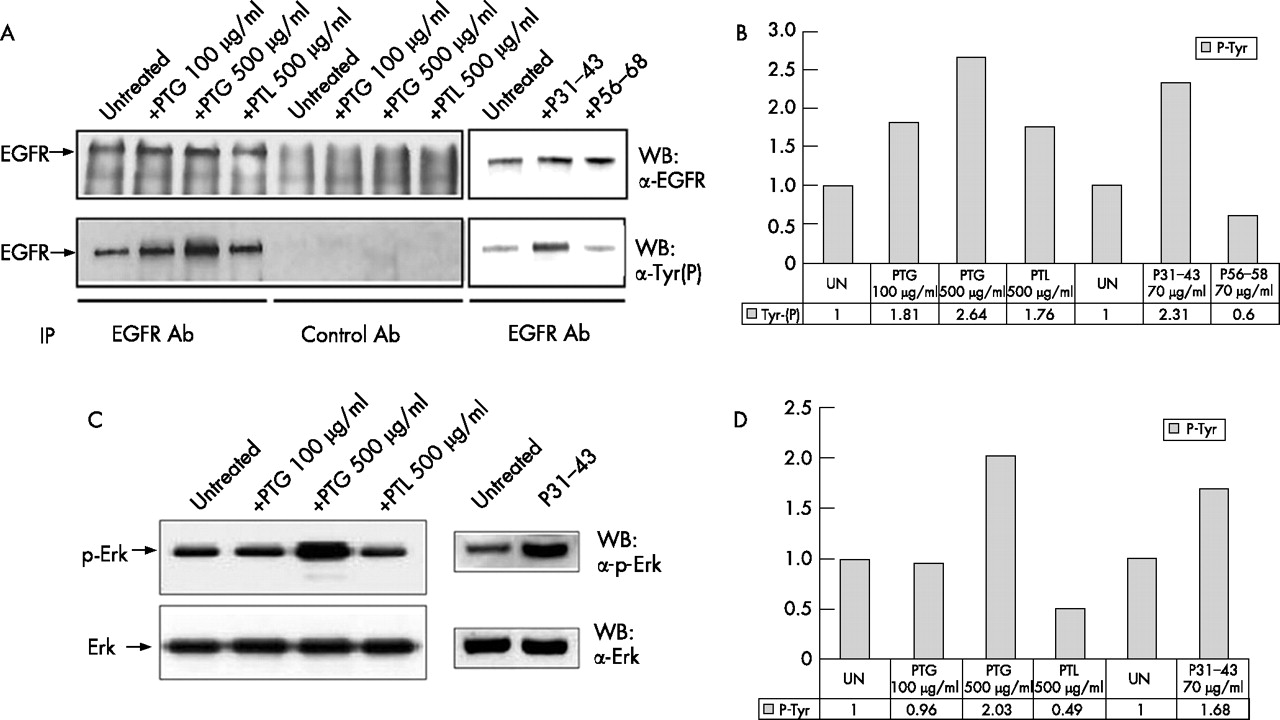
Next we investigated whether gliadin peptides could by themselves induce EGFR activation. As shown in fig 2A⇓, Western blot analysis of the immunoprecipitated EGFR showed that the phospho-tyrosine content of the EGFR increases at least two-fold in comparison to untreated controls after 5 minutes treatment with PTG and P31–43, attaining levels comparable to those observed after EGF stimulation;18 PTL did not increase EGFR activity at similar levels. Densitometric analysis confirmed that the EGFR phosphorylated band increased by 2.64 times after PTG (500 μg/ml) treatment and by 2.31 times after P31–43 (70 μg/ml) treatment compared to the untreated sample (fig 2B⇓). Interestingly, the signal initiated by gliadin peptides also involves downstream effectors of the EGFR such as Erk, as Western blot analysis shows PTG-and P31–43 induced Erk phosphorylation (fig 2C⇓) similar to that seen after EGF stimulation.25 Densitometric analysis indicates a 2.03 and 1.68 fold increase of the Erk phosphorylated band after PTG and P31–43 treatment respectively (fig 2D⇓). PTG 100 μg/ml does not increase Erk phosphorylation in 5 min., but a longer treatment time (e.g. 20 min.) can induce an increment of Erk phosphorylation (Supplementary fig 1⇑). These results indicate that gliadin peptides can fully activate the EGFR pathway.
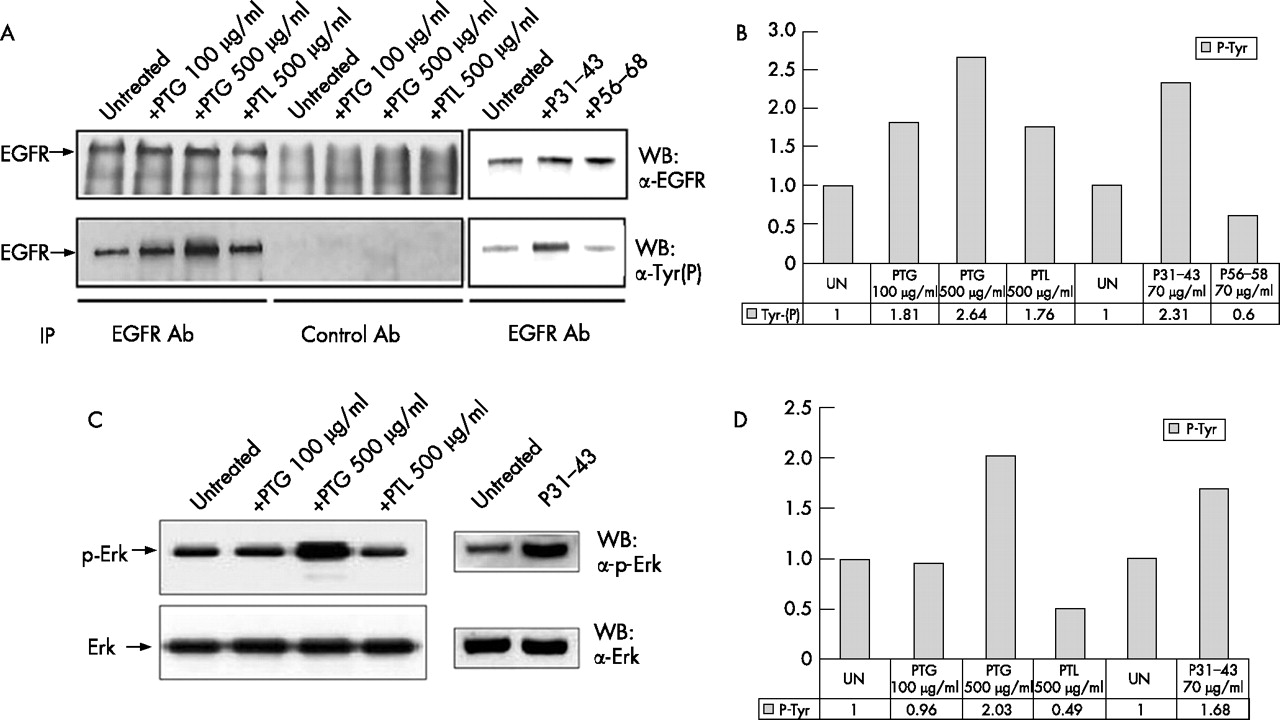
Peptic-tryptic digests of gliadin (PTG) and P31–43 can increase epidermal growth factor receptor (EGFR) and Erk phosphorylation. [A] Immunoprecipitation of the EGFR, with anti-EGFR [upper panel] or anti-phosphotyrosine [lower panel] antibodies shows an increase in EGFR phosphorylation after PTG and P31–43 treatment higher than control lines peptic-tryptic digest of lactoalbumin (PTL), P56–68 and untreated. [B] Densitometric analysis of the Western blot experiment shown in [A]. EGFR phosphorylation is expressed as fold-increase over the corresponding untreated line. UN = untreated. [C] Western blot of Caco-2 total proteins, stained by Erk or p-Erk antibodies shows increased Erk phosphorylation after PTG and P31–43 treatment. [D] Densitometric analysis of the Western blot experiment shown in [C]. Erk phosphorylation is expressed as fold-increase over the corresponding untreated line. UN = untreated. The blots shown are representative of three independent experiments.
Gliadin peptides exert EGF-like effects on G0-S cell cycle transition in NIH3T3
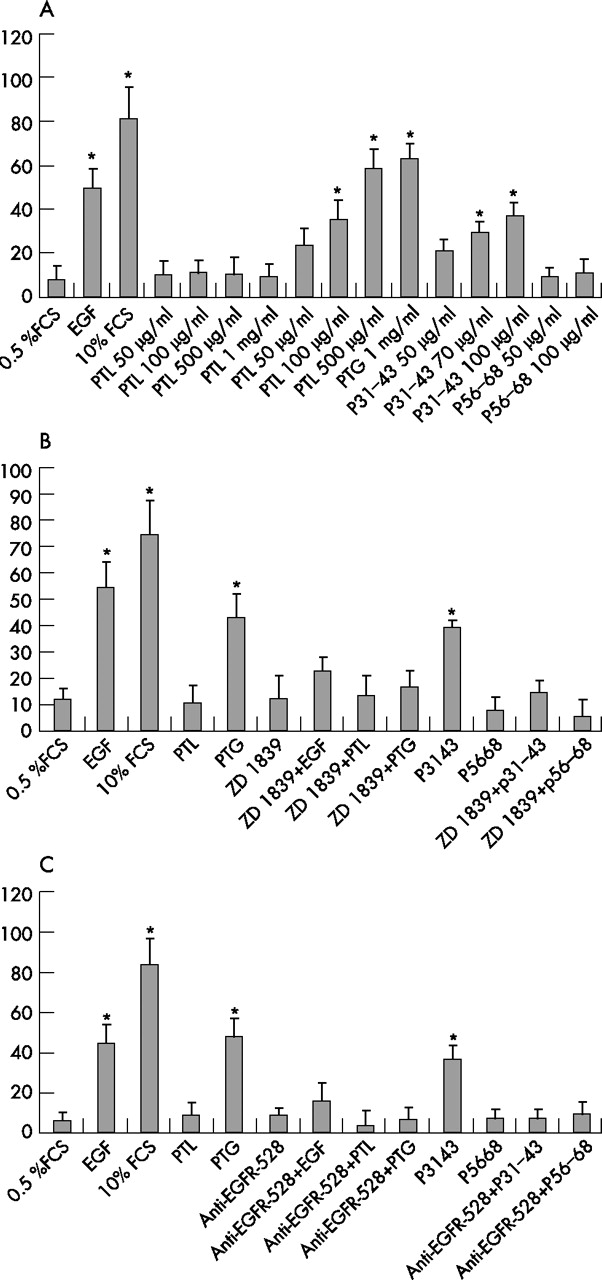
Growth factors such as EGF have a major role in driving G0-S transition in the cell cycle of cells such as resting NIH3T3(Cl7), a cell line generally employed to perform cell cycle experiments as it can be easily synchronised into the G0 phase of the cell cycle. Therefore, if the action of gliadin peptides is tightly linked to the EGF pathway, we predicted that these molecules would have similar effects on DNA synthesis. Since Caco 2 cells cannot be synchronised by serum starvation, it was decided to employ NIH3T3(Cl7) fibroblasts which were synchronised in G0 phase of the cell cycle by serum starvation and treated with gliadin peptides, or with serum or EGF as positive controls. As expected, the addition of serum or EGF results in cell cycle entry of serum starved cells, as determined by increased BrdU incorporation, a marker of S phase (fig 3A⇓). PTG and P31–43 appear to be at least as effective as EGF in driving cells into S phase. PTG and P31–43 induced BrdU incorporation (mean of the percentage) is dependent on concentration and increases up to 59%±8% using 500 μg/ml PTG and to 38%±6% using 100 μg/ml P31–43 (fig 3A⇓).
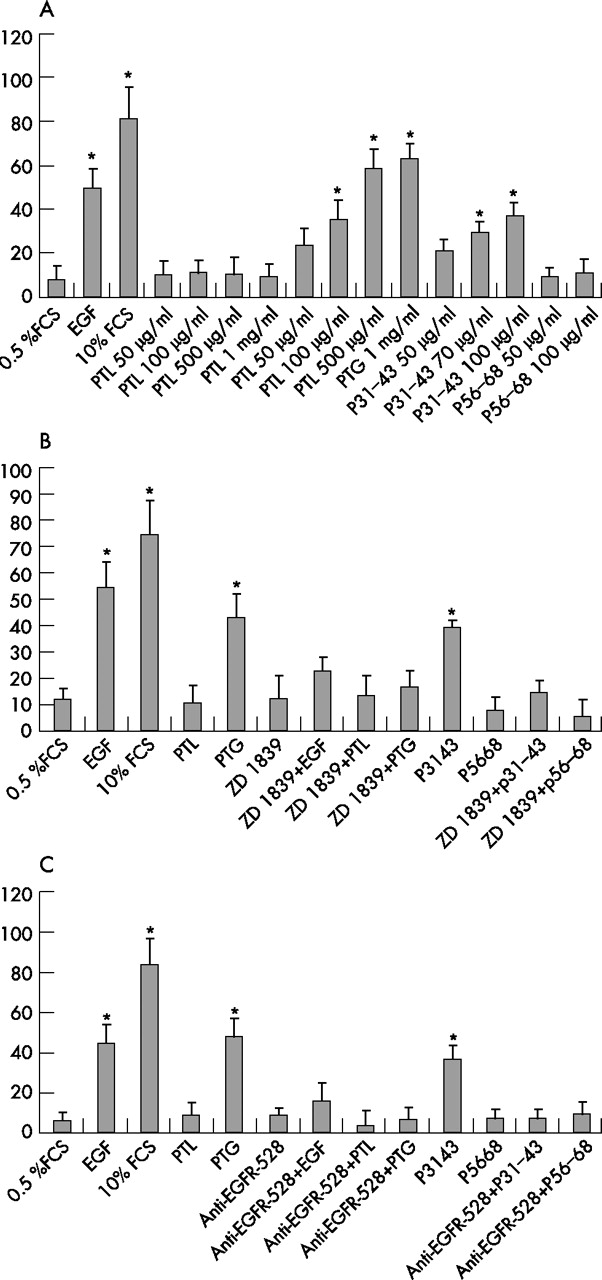
Peptic-tryptic digests of gliadin (PTG) and P31–43 can exert EGF-like effects on cell cycle. [A, B and C] Quantisation of BrdU incorporation in G0 synchronised NIH3T3(Cl7) cells treated as indicated for 18 hours. BrdU was added in all samples for 1 hour before fixation and immunofluorescence staining. The PTG and P31–43 induced G0>S transition of the cell cycle is concentration dependent [A] and can be prevented by epidermal growth factor receptor (EGFR) inhibitors ZD 1839 [B] and anti-EGFR-528 [C]. The bars represent the fraction of BrdU incorporating cells as % of total cells and are the mean + SD of 3 independent experiments. Asterisks indicate P<0.05.
The effects of EGF and gliadin peptides on cell cycle entry depend on EGF receptor activation, as they are all prevented by treatment of starved NIH3T3(Cl7) cells with ZD1839 or anti-EGFR antibody (fig 3B, 3C⇑⇑).
Gliadin peptides are not ligands for the EGFR, but interfere with EGFR endocytosis
The fact that gliadin peptides have similar biological effects to EGF and use the same signal transduction pathway as the EGFR might suggest that gliadin peptides could act as EGF-related functional agonists. Data bank searches revealed no obvious similarities between the P31–43 peptide or gliadin itself with any known ligand for the EGFR or any other receptor. Moreover, suboptimal concentrations of PTG or P31–43 and EGF do not have additive effects in stimulating G0>S transition in the cell cycle, as would be expected if gliadin peptides and EGF were interacting with the same receptor. On the contrary, (fig 4A⇓) low levels of EGF and P31–43 produce a strong synergistic effect, inducing levels of BrdU incorporation comparable to those found after stimulation with optimal concentrations of each agent alone.

Gliadin peptides interfere with epidermal growth factor receptor (EGFR) decay in Caco-2 cells. [A] Suboptimal concentrations of epidermal growth factor (EGF) (10 ng/ml) and P31–43 (10 μg/ml) have synergistic effects on BrdU incorporation by NIH3T3(Cl7) fibroblasts. BrdU incorporation was performed and expressed as described above. Bars represent the mean +/−SD of three independent experiments. Asterisks indicate P<0.05 for T-test. [B] Pulse-chase determination of EGFR activity. Western blot of immunoprecipitated EGFR from Caco-2 cells which were pulsed for 1 h with epidermal growth factor (EGF) at 0°C and chased for the indicated times after temperature shift to 37°C. In PTG and P31–43 treated samples, the EGFR is still activated 90′ after temperature shift, by which time all activated EGFR has been degraded in control cells. [C] Densitometric analysis of Western blot experiments shown in [B]. EGFR phosphorylation is expressed as fold-increase over the corresponding untreated line. UN = untreated. [D]. Endocytic vesicles containing EGF-Alexa488 persist longer after P31–43 treatment of Caco-2 cells. Cells were pulsed for 1 h at 0°C with EGF-Alexa488 and chased after temperature shift to 37°C, with P31–43 or control P56–68 for the times indicated. 63× objective. Representative results from 3 independent experiments.
An alternative possibility to explain the induction of EGFR responsiveness by gliadin peptides could be interference with the decay of the activated EGFR. We studied this by following the activation of the EGFR after endocytosis.19,20 After membrane loading of EGFR with EGF at 4°C, a temperature shift to 37°C initiates endocytosis of the auto-phosphorylated ligand loaded receptor.19,20 As shown by Western blotting, 40 min. later, phosphorylation levels of the internalised EGFR begin to be reduced and become virtually zero at 90 min. (fig 4B⇑, C). In the presence of PTG or P31–43, the receptor is still phosphorylated 90′ after internalisation, indicating significantly extended EGFR activity. In parallel, EGF shows prolonged persistence in endocytic vesicles, as the EGFR still carries EGF-Alexa488 in P31–43 and PTG (not shown) treated cells 90 min. after internalisation, but not in controls (fig 4D⇑).
Gliadin peptides delay EGF-Alexa 488 trafficking in cultured intestinal biopsies from coeliac patients and induce an EGFR-dependent increase in growth rate
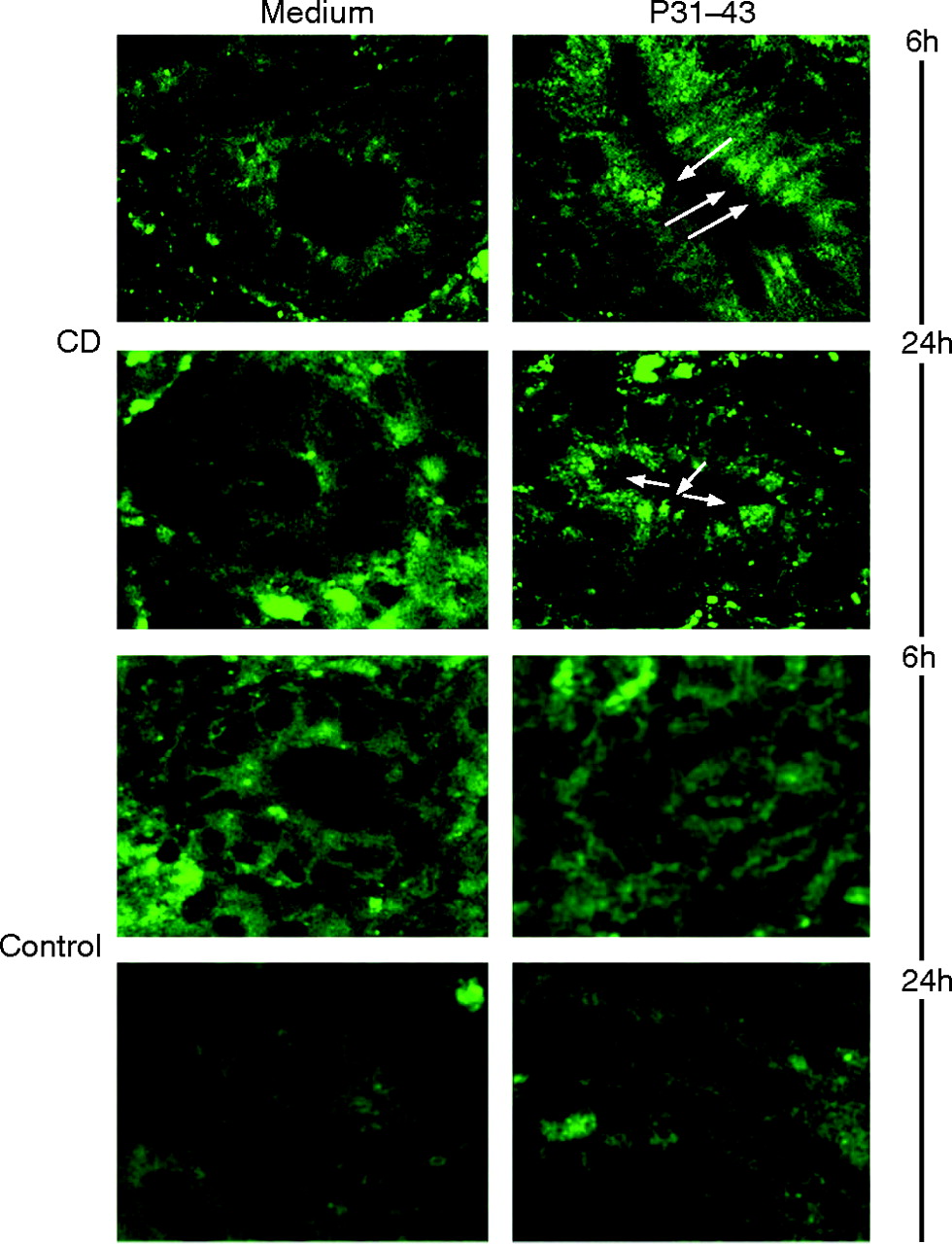
To test the relationship between the EGF-like effects of gliadin and the pathogenesis of CD, pulse-chase experiments examining the fate of EGF-Alexa488 were carried out in biopsies from CD patients with active disease. Biopsies were treated with EGF-Alexa488, washed, and PTG or P31-43 was added for 6 h and 24 h. As shown in fig 5⇓, EGF-Alexa488 can be seen to be localised in endocytic vesicles in the enterocytes of the atrophic villi (Supplentary fig 2⇑) and of the crypts. These labelled vesicles were more apparent in samples treated with either P31–43 (fig 5⇓) or PTG (data not shown) respect to untreated samples, indicating a delay in processing of the EGF carrying vesicles in presence of gliadin peptides in biopsies from untreated CD patients, but not in controls (fig 5⇓).

Persistence of endocytic vesicles containing epidermal growth factor (EGF)-Alexa488 after P31–43 treatment in cultured biopsies from coeliac disease (CD) patients. Duodenal biopsies from CD patients with active disease and controls were pulsed with EGF-Alexa488 for 3 h and chased for 6 h and 24 h in the presence of P31–43 or with medium alone [Medium]. White arrows indicate accumulation of EGF-Alexa488 carrying vesicles in the epithelial cells in the P31–43 treated sample. 63× objective. The figure shows one representative experiment from 3 independent experiments.
The persistence of EGF in endocytic vesicles raises the possibility that prolonged activity of the EGFR could account for some of the typical alterations in the CD mucosa, such as increased crypt cell proliferation. Treatment of these biopsies with PTG (500 μg/ml) or P31–43 (70 μg/ml) increases the proportion of BrdU incorporating cytokeratin expressing epithelial cells to 43%±5% (mean and SD) and 37%±8% (mean and SD) respectively compared with 8%±6% (mean and SD) of epithelial cells in samples cultured in the absence of gliadin. Treatment with anti-EGFR antibody reduces the gliadin-induced increase in BrdU incorporation to 12%±5% for PTG and 10%±8% for P31–43 (fig 6A⇓, B). The control reagents PTL and P56–68 have no effect on BrdU incorporation in these assays (not shown). Consistent with earlier studies,14,21 culture of biopsies from untreated CD patients with PTG and P31–43 causes an increase in the fraction of apoptotic epithelial cells as assessed by the TUNEL assay, compared to the controls cultured with medium alone (fig 6C⇓). However, EGFR activation appears to play no role in the apoptotic effect of gliadin peptides, as EGFR inhibitors fail to prevent gliadin-induced apoptosis of enterocytes in the cultured intestinal biopsies (fig 6C⇓). Neither increase of BrdU incorporation nor tunnel positive cells could be detected in cultured biopsies from non-coeliac patients treated with PTG and P31–43 (fig 6⇓ B, C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Blocking the epidermal growth factor receptor (EGFR) prevents gliadin induced S-phase entry of enterocytes in cultured biopsies from coeliac disease (CD) patients. [A] Duodenal biopsies from patients with active CD were stained with cytokeratin to identify epithelial cells [red] and for BrdU [green]. [B] Quantisation of BrdU incorporation of intestinal biopsies from CD patients and controls; more than 300 cytokeratin positive cells were counted in several fields in each sample and the number of BrdU positive cells was expressed as a proportion of the total of cytokeratin positive cells. [C] The percentage of apoptotic epithelial cells was calculated by determining the number of TUNEL positive cells with respect to the total number of cytokeratin positive cells. Both in [B] and [C] the results shown are the mean and standard deviation. The numbers in the columns are the numbers of the samples for each point. 63× objective. Asterisks indicate P<0.05.
DISCUSSION
In this study we have shown that gliadin peptides induce actin rearrangements and cell proliferation in a wide range of cell types, mimicking the effect of EGF. The EGF pathway was in fact enhanced by gliadin, this phenomenon being due to delayed inactivation of EGF receptor. The effect was also present in a more complex system represented by the cultured small intestinal mucosa from patients with untreated CD. We think that these observations add a new biological function to gliadin peptides in addition to their ability to activate the innate and adaptive immune response, which is in
any case related to the role these proteins play in the remodelling of the coeliac mucosa. We have confirmed11 the ability of PTG and P31–43 to induce actin rearrangements in epithelial cell lines of intestinal origin such as Caco-2, but also in other cell lines. They can affect cells from a variety of different origins such as MCF7 cells, an epithelial cell line from a human mammary carcinoma and mouse skin fibroblasts NIH3T3(CL7)(Cl7) (data not shown). They act very rapidly (10–15 min.) and they produce similar morphological changes in the cell, leading to highly characteristic membrane ruffling. The effect was strongly reminiscent of that induced by growth factors. Of the several growth factors we tested, including PDGF, insulin and LPA (data not shown), only EGF could mimic the effects of gliadin peptides on Caco2 cells. The involvement of EGF was consistent with the high expression of EGFR on these cell lines.15,17,18
The rapid EGF-like effects of gliadin peptides on the actin cytoskeleton prompted us to search for further evidence of EGFR pathway involvement. Like EGF, PTG and P31–43 induced G0->S transition in resting NIH3T3(Cl7)[l5] cells, confirming the hypothesis that gliadin peptides share other effects of EGF in addition to cytoskeletal modifications. The direct involvement of the EGFR pathway was proved by the ability of inhibitors such as ZD1832 and anti-EGFR blocking antibody to prevent the effects of gliadin peptides. In parallel, the peptides induced phosphorylation of the EGFR and of the downstream effector signalling molecule Erk, indicating activation of the EGFR pathway.
How do gliadin peptides elicit their EGF-like effects? PTG and P31–43 are not known to bind the EGFR, and we could not find any sequence similarity with EGF or any other growth factor, suggesting they do not act as direct ligands for the EGFR. Moreover, suboptimal concentrations of P31–43 and EGF showed a clear synergistic effect on S phase entry, rather than the additive effect which would be expected if they interacted with the same receptor. Growth factor receptor activity can be regulated by ligand binding, but also by interference with degradation of activated receptors.19,20 Endocytosis and receptor inactivation were indeed delayed in PTG and P31–43 treated cells, with activated EGFR still being present 90 minutes after temperature shift, a time point when inactivation was complete in untreated cells. Moreover, gliadin peptides interfered with the trafficking of vesicles carrying EGF-Alexa488. Although little is known about the viability of gliadin peptides, there are indications that they enter the enterocytes.26–,28 Experiments conducted employing biopsy specimens from untreated coeliac patients mounted in Ussing chambers, indicate transcellular transport of the peptide 31–49.26 Furthermore, both an antibody that recognises a large gliadin peptide P3–56 and an anti-gliadin antibody stain the Golgi and the vesicular compartment in intestinal biopsies from CD patients on a gluten containing diet studied by electron microscopy and immunofluorescent techniques.27,28 Data from our laboratory suggest that gliadin peptides P31–43 and P56–68 labelled with fluorochromes enter Caco2 cells and interact with the vesicular compartment.
Increased crypt epithelial cell proliferation is a highly characteristic feature of the damaged mucosa in CD,21 a phenomenon generally thought to be secondary to an antigen-specific mucosal immune response against gliadin.29 However, the data presented here show a further, not completely alternative, explanation, according to which gliadin peptides are able to enhance proliferation in the coeliac mucosa enterocyte, to delay the trafficking and degradation of EGFR, and to enhance the sensitivity of epithelial cells to the effects of endogenous EGF, which is the most potent known mitogen for epithelial cells.25 This idea is supported by our pulse-chase experiments with Alexa488 labelled EGF in mucosa from untreated patients in active phase of the disease, which showed a delay of trafficking of the EGF carrying vesicles in epithelial cells after treatment with P31–43. In addition, the increased epithelial cell proliferation rate induced by gliadin peptides was prevented by a blocking anti-EGFR antibody. Interestingly, these effects of gliadin peptides are specific for proliferation, as gliadin induced apoptosis of epithelial cells in cultured biopsies from untreated patients was not prevented by the EGFR inhibitors. Different receptors, such as Fas, are probably mediating apoptosis in this case.21 Altogether these observations suggest that the EGF pathway plays a central role in initiating and maintaining the high proliferation rates observed in the crypts of patients with CD. The evidence described here offers a novel explanation for the histological features of the CD mucosa, in which persistent epithelial cell proliferation leads to inhibited maturation and differentiation of epithelial cells and loss of the normal villous structure.
The effects of gliadin peptides on epithelial cell proliferation we observed in CD mucosa were too rapid to be secondary to T-cell mediated recognition of gliadin. However, the increased sensitivity to EGFR signalling induced by gliadin peptides might act in synergy with other effects of gliadin on early components of the innate immune response, or even of the adaptive immune response.29–,31 The early immune response to gliadin peptides, particularly to P31–43 or P31–49 peptides, includes epithelial expression of MIC-A, an effect relayed by IL15,32 and epithelial phosphorylation.33 Data presented here raise the possibility that some gliadin effects on the immune or adaptive response could reflect enhancement of the EGFR signalling pathway in epithelial cells. It is interesting in this context to note that the signal transduction pathways of IL15 are shared by growth signals induced by EGF.34
The complex entanglement of genetic and environmental factors in CD will be the key to explain the specificity of gliadin effects on coeliac mucosa. One working hypothesis could be that an alteration of some genetic factor35 in coeliac patients may lead to a deregulated activity of the endocytotic pathway that is apparently compensated in the absence of gliadin. Acting on the same pathway, either directly or indirectly through the innate immunity, gliadin can induce the alterations of the CD intestinal mucosa. Gliadin could therefore be the necessary trigger to turn a somehow tolerated, but already altered cell state, into an evidently pathological one.
REFERENCES
Footnotes
Funding: The project was financed by Regione Campania (legge 5) and MIUR (PRIN).