Article Text

Abstract
Background and aims: Recurrent pancreatitis is a common complication of severe hypertriglyceridaemia in patients with various gene mutations in lipoprotein lipase (LPL) or apolipoprotein CII. However, the exact pathogenetic mechanism has not yet been defined.
Methods: Susceptibility to pancreatitis in LPL-deficient mice was compared with that of wild-type mice after intraperitoneal injections of caerulein by determination of amylase release and pancreatic pathological scores. The effect of chylomicrons and fatty acids on enzyme release, Ca2+ signalling and cell injury in isolated pancreatic acinar cells from wild-type and LPL-deficient mice was investigated.
Results: Caerulein induced higher levels of serum amylase and more severe inflammation in the pancreas of LPL-deficient mice than in wild-type mice. Addition of free fatty acids or chylomicrons to isolated pancreatic acinar cells led to the release of amylase and caused cell injury at higher concentrations. The effect of chylomicrons was partially blocked by orlistat, an inhibitor of pancreatic lipase. These results suggest that increased concentrations of free fatty acids from chylomicron hydrolysis by pancreatic lipase can induce acinar cell injury. Surprisingly, pancreatic lipase, whether in its active or inactive state could act like an agonist by inducing amylase secretion without cell injury. It caused an increase in cGMP levels and conversion of cell-damaging sustained elevations of [Ca2+] to normal Ca2+ oscillations.
Conclusions: LPL-deficient mice with severe hypertriglyceridaemia display enhanced susceptibility to acute pancreatitis. High levels of chylomicrons and free fatty acids result in pancreatic cell injury. Pancreatic lipase has a dual effect: generating free fatty acids from chylomicrons and preventing Ca2+ overload in pancreatic acinar cells.
Statistics from Altmetric.com
Although the pathogenesis of acute pancreatitis is multifactorial, its recurrent form is the most common complication of severe hypertriglyceridaemia (HTG) often seen in type I hyperlipoproteinaemic patients with various gene mutations in lipoprotein lipase (LPL) or apolipoprotein CII.1 It was hypothesised that in severe HTG pancreatic lipase seeping from pancreatic acinar cells to the interstitial cell side hydrolyses triglycerides (TGs) in chylomicrons (CMs) and very low density lipoproteins. This would generate free fatty acids (FFAs), resulting in pancreatic cell injury.2 3 Although TGs in CMs are the natural substrate for LPL, TGs in CMs are also hydrolysed by purified pancreatic lipase.4 FFAs have been reported to induce activation of trypsinogen and to initiate acute pancreatitis,5 which is aggravated by alcohol consumption and formation of fatty acid ethyl esters6 7 with progression to chronic pancreatitis. Furthermore, abnormal cytosolic calcium release has been found to induce trypsinogen activation8 9 and acinar cell damage.10 11 Conversion of the normal oscillations into abnormal sustained elevations of cytosolic Ca2+ concentrations appears to play a key role in the pathogenesis of acute pancreatitis. In seeking the causal relationship of HTG and acute pancreatitis, various animal models have been investigated.12 13 Recently, we successfully rescued homozygous LPL gene-targeted mice by somatic gene transfer of a mutant LPL gene. The animals that survived exhibited LPL deficiency with extremely high plasma triglyceride levels during adulthood.14 The present study was therefore carried out to assess the susceptibility to pancreatitis in these LPL-deficient mice with severe HTG. Furthermore, isolated pancreatic acinar cells were incubated with CMs derived from blood plasma of these LPL-deficient mice to test the role of pancreatic lipase, high TG and FFA levels, and Ca2+ signalling in the development of cell injury and pancreatitis. We found that elevated TGs and FFAs are high risk factors but an additional stimulus that leads to toxic effects such as high and long lasting levels of cytosolic [Ca2+] is necessary to evoke damage of isolated pancreatic acinar cells. Surprisingly, pancreatic lipase, whether in its active or inactive state, can act in a receptor-dependent manner when reaching the interstitial cell side. It converts damaging sustained elevations of [Ca2+] to more normal Ca2+ oscillations. This indicates that in addition to its enzymatic activity, which leads to hydrolysis of TGs from CMs, pancreatic lipase can also play a protective role which counteracts cell injury.
MATERIALS AND METHODS
Detailed methods can be found in the supplementary data. Female wild-type and rescued LPL-deficient mice were used for experiments. Genotyping was performed by polymerase chain reaction. Pancreatitis was induced by intraperitoneal injections of caerulein (20 μg/kg). The pancreas was sectioned and the degree of pathological alteration was scored by histological inspection. CMs were isolated from blood plasma of LPL-deficient mice by ultracentrifugation. Lactate dehydrogenase (LDH) activity and the concentrations of amylase, lipase, TGs, cGMP and cAMP were determined with commercial kits. FFAs and free cytosolic [Ca2+] were determined with fluorescence indicators. Production of the active human pancreatic lipase (HPL), the inactive lipase mutant (HPL S152G) and the inactive lipase-related protein1 (HPLRP1) were produced in the yeast Pichia pastoris. Pancreatic lipase (>20 000 U/mg protein) was obtained from Sigma Aldrich, Steinheim, Germany (cat. no. LO382). The results are expressed as mean with the SD. Statistical significance was determined by analysis of the variance (ANOVA) and p values <0.05 were regarded as significant.
RESULTS AND DISCUSSION
LPL-deficient mice with severe HTG have enhanced susceptibility to pancreatitis
Since LPL is localised in the endothelia of capillaries and is therefore present in pancreatic tissue but not in isolated acinar cells, susceptibility to pancreatitis was tested in intact pancreatic tissue. There were no obvious histological differences in the pancreas of wild-type and LPL-deficient mice injected with saline. However, mice injected with caerulein exhibited pancreatic injury including oedema, neutrophil infiltration, haemorrhage and parenchymal necrosis. In LPL-deficient mice, pancreatic injury was more severe than in wild-type mice, as illustrated in fig 1. Using morphological scores for semi-quantitative estimation of the degree of pancreatic injury, the total histological scores in LPL-deficient mice were significantly higher (16.17 (SD 2.14) vs 11.00 (SD 1.85), see table 1). This was mainly due to increased scores in haemorrhage and necrosis (4.00 (SD 1.10) vs 1.63 (SD 0.74) and 4.17 (SD 0.41) vs 2.75 (SD 0.46), respectively; see table 1). Although neutrophil infiltration (inflammatory infiltration) and oedema scores were increased by 24% and 17% in LPL deficient mice, they failed to reach statistical significance.
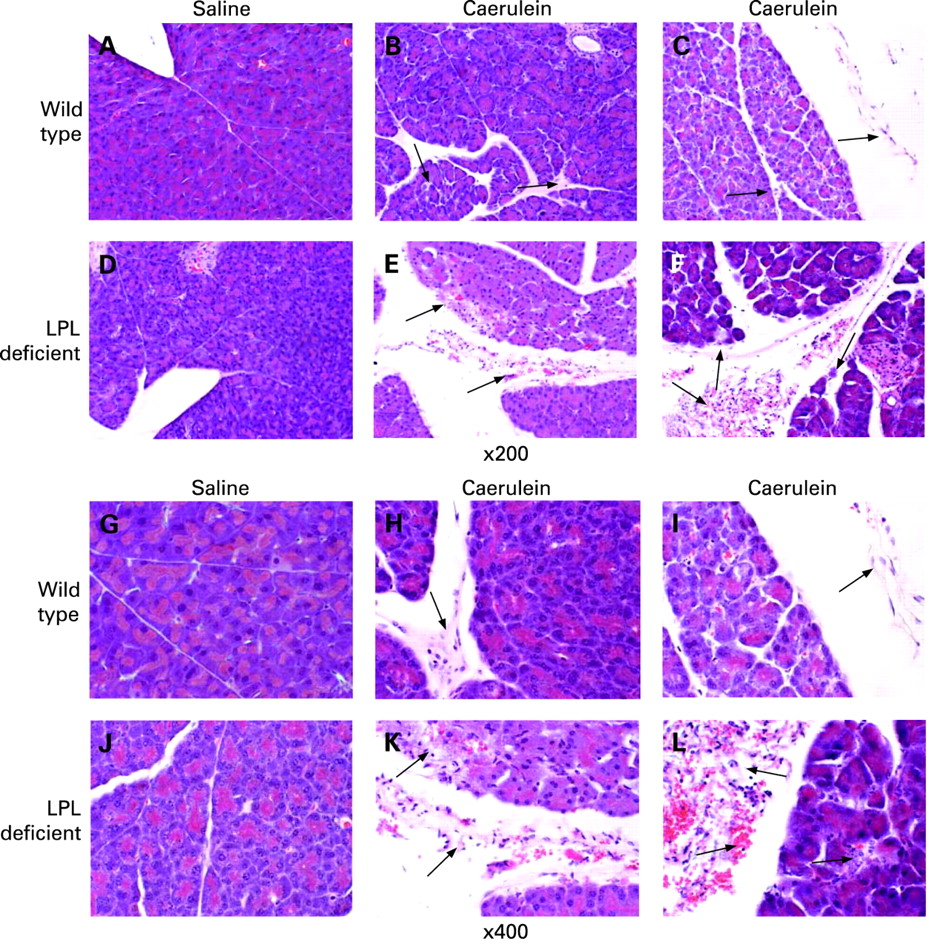
Plasma triglyceride levels and histological scores for pancreatic injury
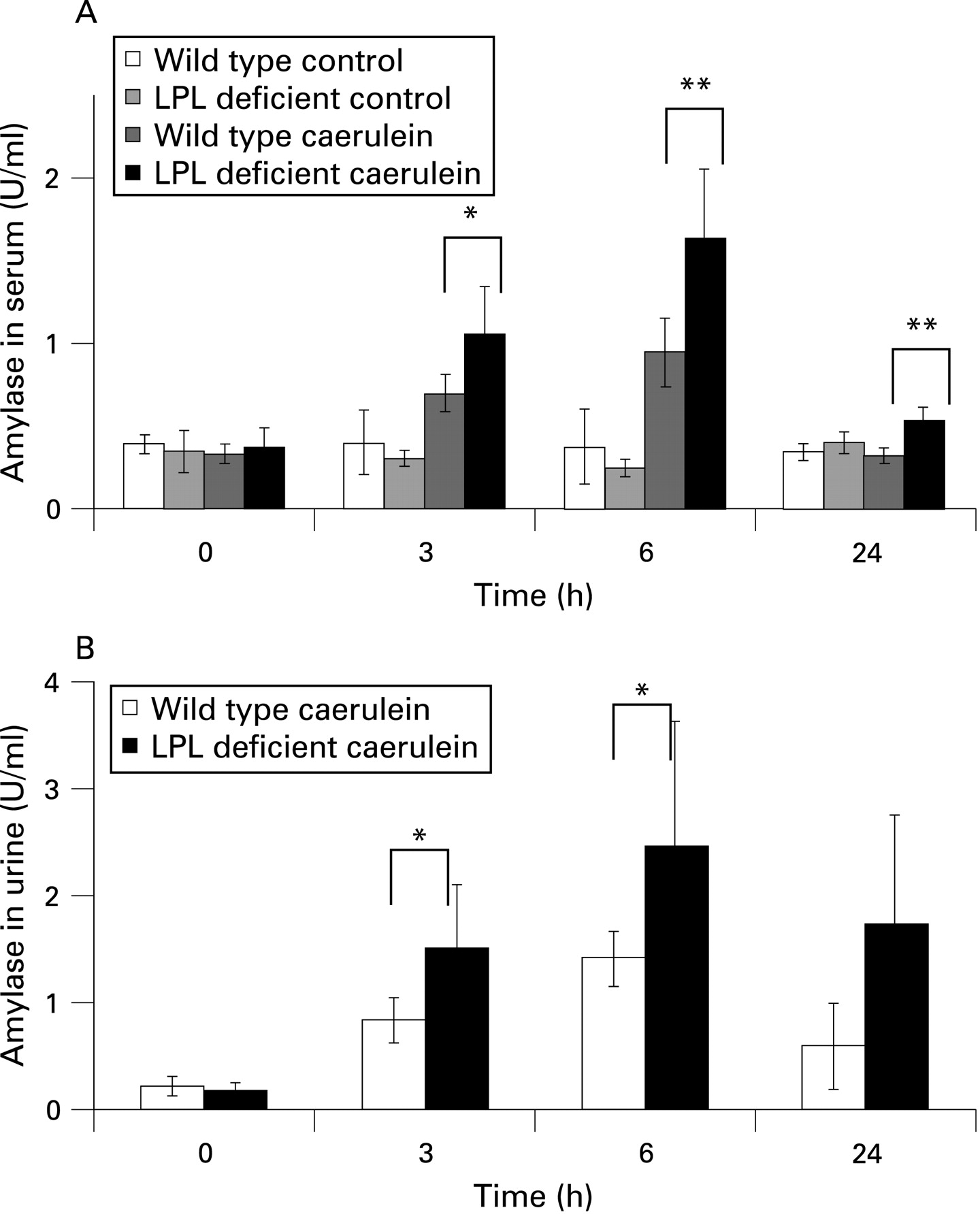
Serum and urine amylase levels increased after injection of caerulein in both wild-type and LPL-deficient mice, but were significantly higher in LPL-deficient mice 3 and 6 h after injection (fig 2A,B; p<0.05 and p<0.01) There was no discernible pathological alteration in either wild-type or LPL-deficient mice injected with saline as a control.

We also examined whether hyperlipidaemic conditions could act as a sensitiser for the development of pancreatitis at very low doses of caerulein (0.1 µg/kg and 0.5 µg/kg body weight), in a similar manner to that described in ethanol-fed animals.15 However, injections of these low doses resulted neither in increased plasma amylase levels nor in morphological alterations in the pancreas of either wild-type or LPL-deficient mice, indicating that HTG does not sensitise LPL-deficient mice to pancreatitis caused by caerulein (see supplementary figs 1 and 2). Furthermore, we investigated whether lack of LPL rather than severe HTG alters susceptibility to pancreatitis. We have therefore prepared isolated acinar cells from neonatal pups instead from adult mice in order to avoid the influence of severe HTG. Although addition of cholecystokinin (CCK) (10 pmol/l) or chylomicrons (10 mg/ml) or a combination of both could induce significant release of amylase and lipase from acinar cells of neonatal pups, no difference could be observed between wild-type and LPL-deficient neonatal mice (see supplementary fig 3).

Free fatty acids are generated from chylomicrons and lead to pancreatic acinar cell damage
Using pancreatic acini from wild-type mice, and CMs from the blood plasma of LPL-deficient mice, we have tested the hypothesis that pancreatic lipase seeping from pancreatic acinar cells leads to generation of FFAs from triglycerides of CMs.
As shown in fig 3A,B the release of FFAs into the medium increased with time following addition of CCK or commercial pancreatic lipase. The increase was higher in the presence of CMs at concentrations found in LPL-deficient mice (table 1) and in patients with HTG.2 Small amounts of FFAs were generated if cells were incubated without any additions for 25 min (see fig 3A, control). When cells were stimulated with CCK in the absence of CMs, a delayed release of FFAs was detectable whose source could be triglycerides that had remained in the cells. Accelerated release of FFAs could be due to CCK-stimulated pancreatic lipase secretion.
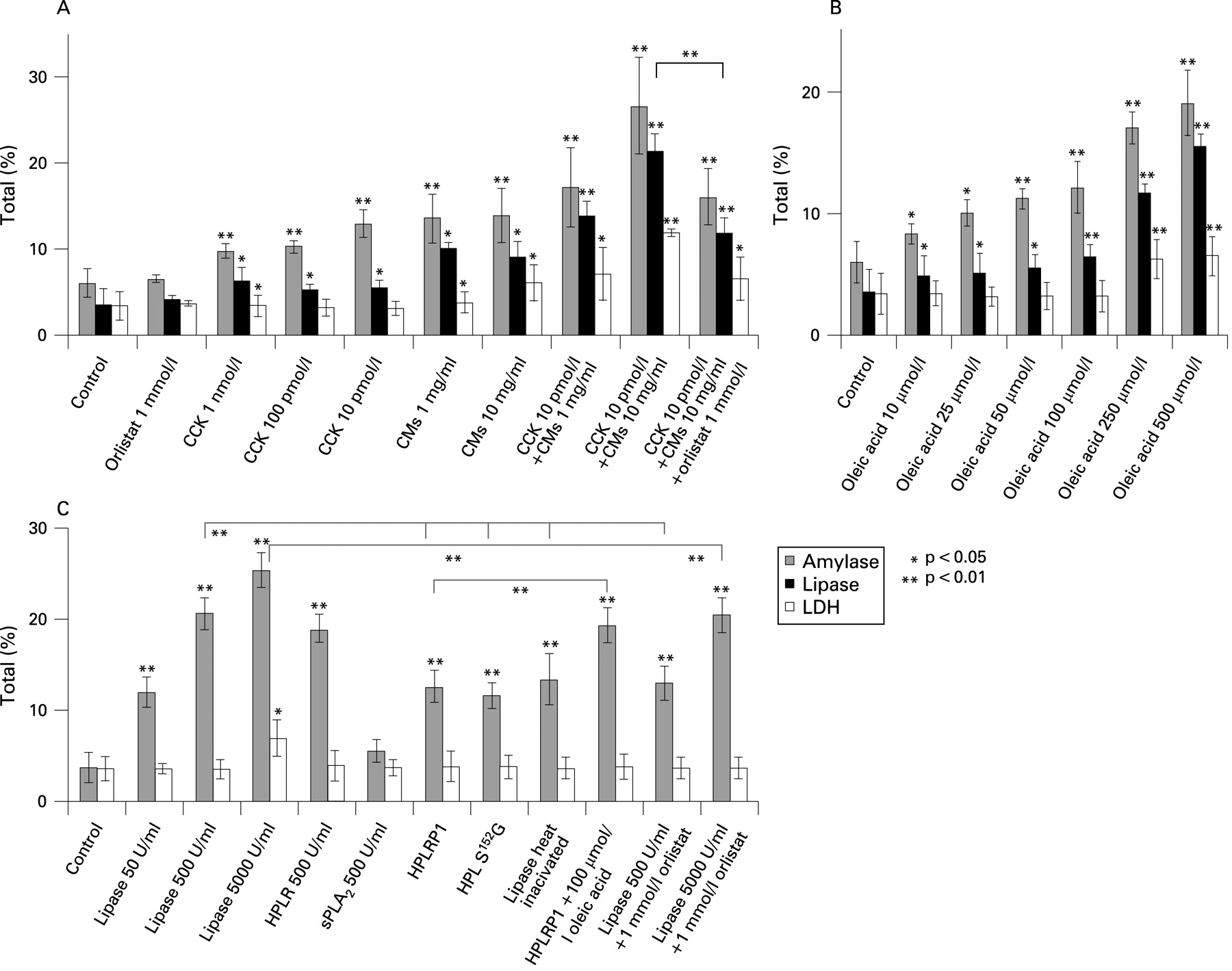
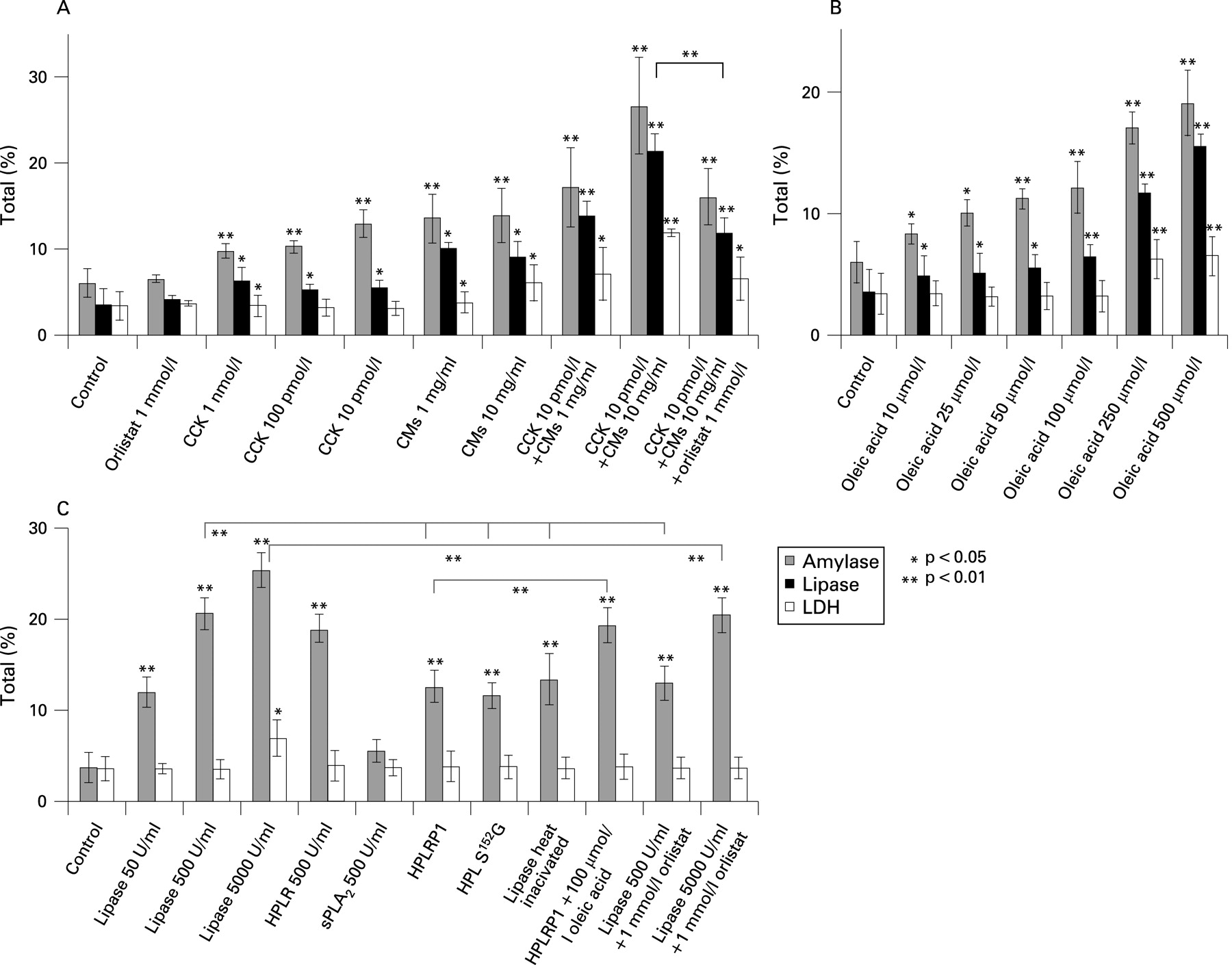
Cell-associated FFA generation was markedly increased in the presence of CMs (10 mg/ml) with or without CCK after 30 min of incubation (fig 3C). Addition of CMs or an FFA (oleic acid C18:1; Sigma) to isolated cells led to enzyme release (fig 4A,B). In combination with CCK, CMs (1 mg/ml) and FFAs (0.1–0.9 mmol/l) at concentrations which are considered normal in healthy individuals16 17 were already toxic if they came into direct contact with cells as in our test system. At these concentrations the release of LDH, as an indicator for cell damage, occurred (fig 4A,B).
Pancreatic lipase hydrolyses triglycerides in chylomicrons
The concentration of TGs in CMs (1 mg/ml) decreased by 15% during incubation for 30 min in the presence of cells whereas the TG concentration remained stable in the absence of cells (fig 3D). Addition of CCK (10 pmol/l, Sigma) accelerated TG breakdown to 25% after 30 min probably due to CCK-stimulated release of pancreatic lipase. In the absence of CMs no TG breakdown was observed within 30 min whereas TG concentration was reduced by 20% in the presence of CCK even in the absence of CMs due to the TGs present in cells (see fig 3D bottom and expanded graph in fig 3E). Interestingly, we found that TGs of CMs were also hydrolysed by added pancreatic lipase (fig 3D). We suppose it is endogenous pancreatic lipase released from pancreatic acinar cells that breaks down TGs in CMs in our experimental system. CMs and a FFA (oleic acid, C18:1) directly added to cells at concentrations >1 mg/ml and >100 μmol/l, respectively, led to cell damage indicated by increased LDH release (fig 4A,B). It is probably FFAs released from CMs (see fig 3A) which play the important role in pancreatitis associated with HTG.
CMs and CCK stimulate the release of amylase, lipase and LDH in isolated acinar cells
Stimulation of pancreatic acinar cells with CCK increased secretion of amylase and lipase by approximately 2-fold of the control cells without hormonal stimulation. As an indicator for cell leakage, release of LDH was not significantly increased after 30 min of stimulation with CCK up to a concentration of 100 pmol/l (fig 4A). However, together with 1 or 10 mg/ml CMs even at 10 pmol/l CCK the increase in both amylase and lipase was about 3- to 5-fold the control, and LDH release was significantly increased up to 4-fold of the control. In this case amylase is certainly not only “secreted” as under hormonal stimulation without LDH release but it is rather a mixture of both “secretion” and “leakage”. The latter will indicate cell damage similar as LDH release. We therefore use the term “release” to indicate conveyance of enzymes from the cell into the surrounding buffer. Cell injury could be reduced by the lipase inhibitor orlistat (Sigma; see fig 4A). Incubation of cells with CMs alone (1 and 10 mg/ml) increased both amylase and lipase release 2- and 3-fold of the control and LDH release by 2-fold the control. In the presence of both CCK and CMs the effect on the release of amylase, lipase and LDH was higher than with each agonist alone. These data indicate that high concentrations of CMs cause cell damage probably due to generation of large amounts of FFAs (see figs 3A and 4B). Cell damage is obviously increased by the release of pancreatic lipase which was higher in the presence of CCK and could be reduced by the lipase inhibitor orlistat (fig 4A).
Effect of free fatty acids on enzyme secretion
In order to investigate the effects of FFAs on pancreatic acinar cells, we added oleic acid (C18:1) to the incubation medium. Release of amylase and lipase was about 3- to 4-fold the control at maximal effective concentrations of oleic acid (0.25 and 0.5 mmol/l; fig 4B). Release of LDH was also significantly increased indicating cell damage at high concentrations of the FFA.
Effect of pancreatic lipase on enzyme secretion
As shown in fig 4C addition of commercial pancreatic lipase (Sigma) to cells (500 U/ml) increased amylase release by approximately 5-fold compared to the control without an increase in the release of LDH. At 5000 U/ml, however, LDH release was also increased, although it could be suppressed to control values in the presence of the lipase inhibitor orlistat (see fig 4C).
Therefore, part of the release of digestive enzymes and LDH in the presence of CMs was due to lipase secreted with consequent generation of FFAs from CMs.
When 170 μg/ml (a concentration which is equivalent to 500 U/ml) of heat-inactivated pancreatic lipase or similar amounts of the inactive lipase mutant (HPL(S152G)) or the lipase-related protein HPLRP1, which lacks enzyme activity, were added to pancreatic acinar cells, amylase release increased the control 3-fold without LDH release (fig 4C). This indicates that part of the lipase effect on amylase release is not coupled to its enzymatic activity. Apparently, the lipase protein is, by itself, able to evoke enzyme secretion. In a separate study we could also show that in the presence of lipase CCK at different concentrations (10−6 mol/l, 10−9 mol/l and 10−12 mol/l) evoked higher amylase release than in its absence. LDH release was even lower in the presence of active or inactive lipase than in its absence (see supplementary figs 4 and 5). We assume that lipase, in addition to its enzymatic activity, also has an agonist function and acts protectively on pancreatic acinar cells (see below). The structurally unrelated pancreatic phospholipase A2 (Sigma), had no effect on amylase release (see fig 4C).
Stimulators of amylase release evoke Ca2+ signalling and cGMP production
[Ca2+]cyt in the presence of chylomicrons and oleic acid
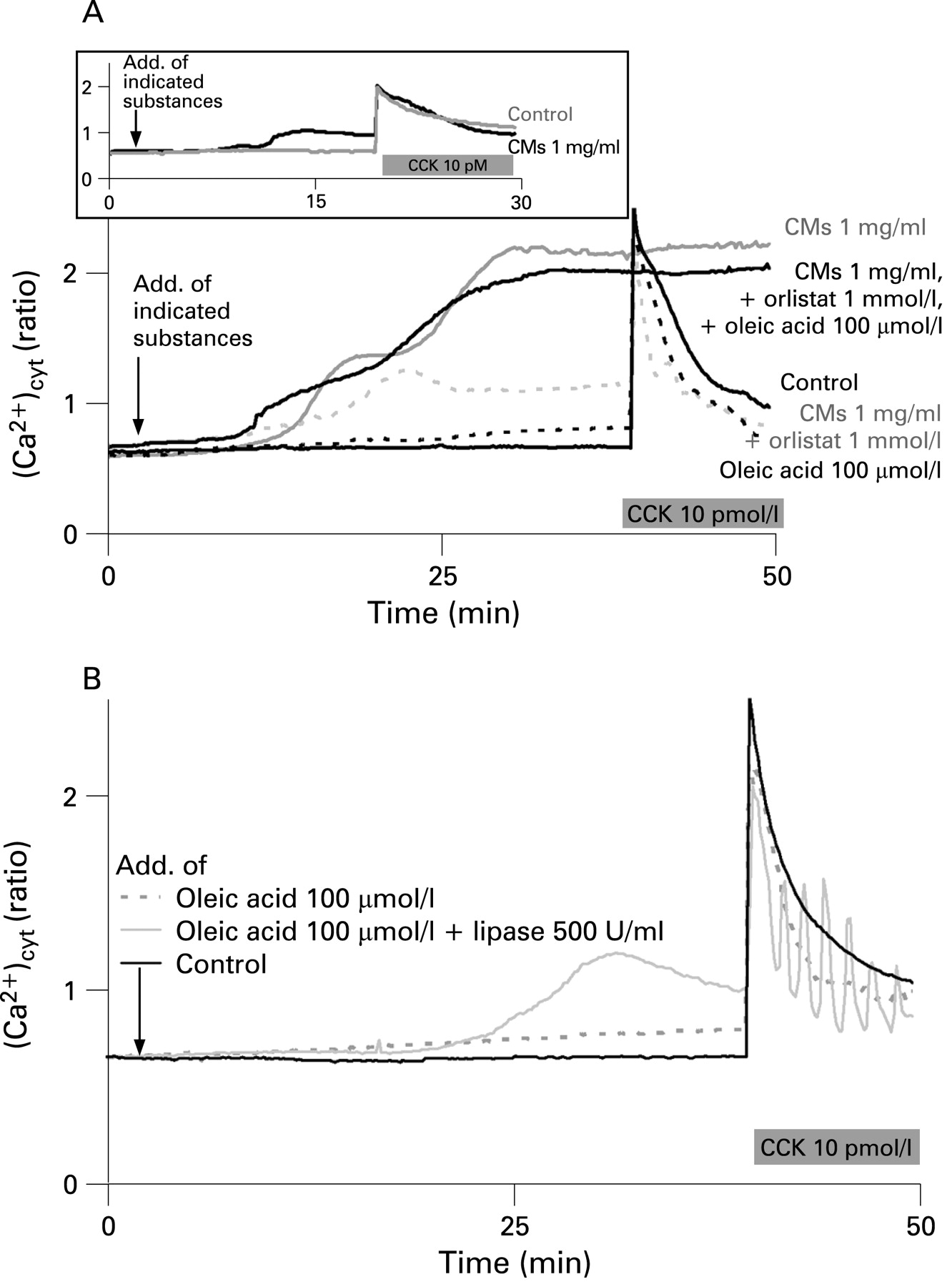
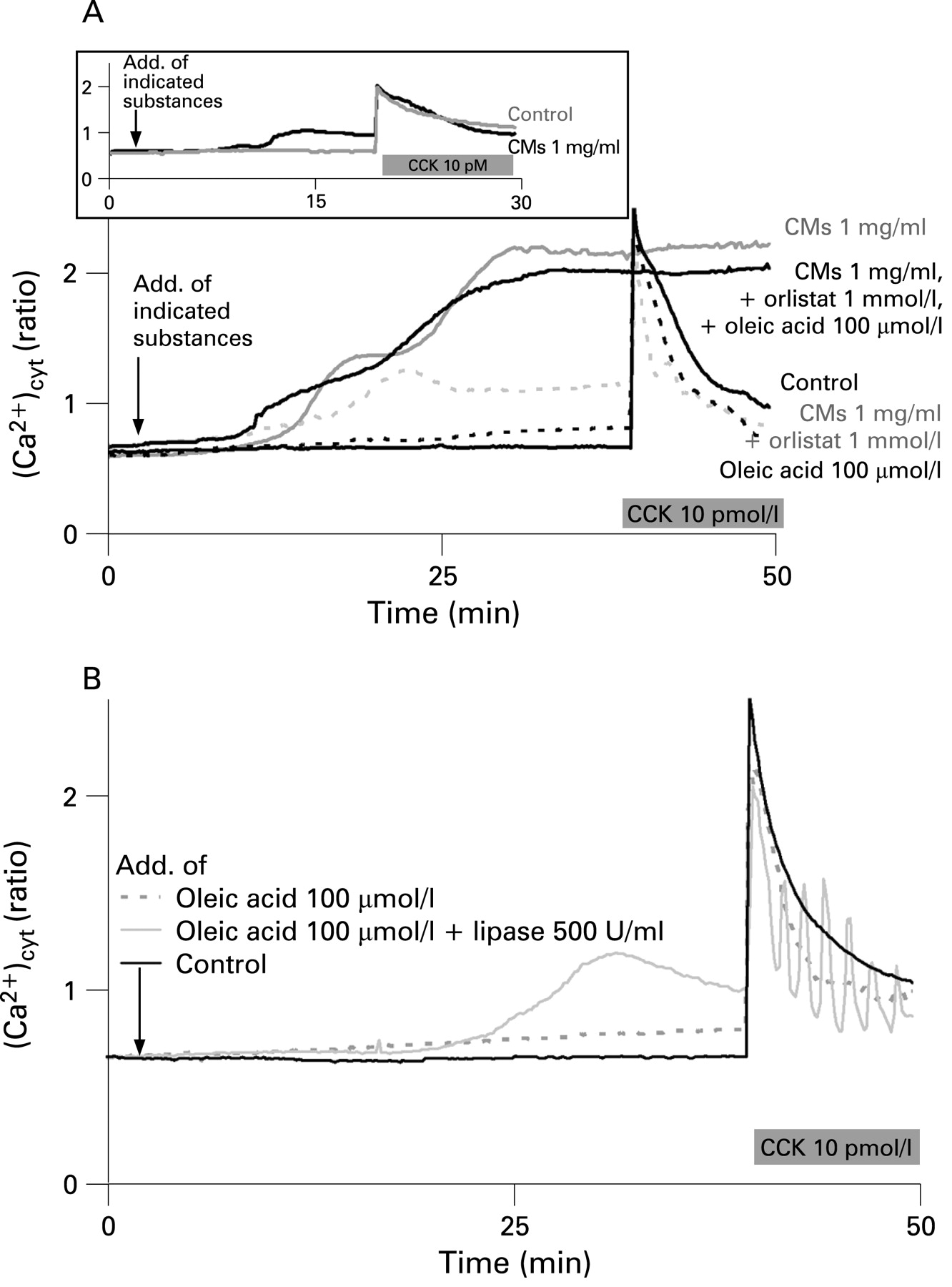
Addition of CMs (1 mg/ml) to cells caused a time-dependent increase in cytosolic Ca2+ concentration until a first plateau of [Ca2+]cyt was reached. Subsequent addition of CCK caused a rapid additional Ca2+ release (fig 5A, upper panel). The initial plateau phase was followed by a second phase of increase in [Ca2+]cyt in the presence of CMs (fig 5A, lower panel). Addition of CCK at the plateau of the second phase did not induce any further increase in [Ca2+]cyt indicating that cells were damaged at this high [Ca2+]cyt. As recently shown by Criddle et al7 fatty acid ethyl esters increase [Ca2+]cyt through inositol trisphosphate receptors. Since fatty acid ethyl esters and fatty acids are known to uncouple oxidative phosphorylation18 failure of the Ca2+ pump function caused by insufficient ATP generation may provide an explanation for the rise of [Ca2+]cyt induced by these metabolites and for the consequent cell necrosis.7 The second phase of CM-induced increase in [Ca2+]cyt was suppressed in the presence of orlistat (1 mmol/l). Subsequent addition of CCK caused a Ca2+ signal that peaked to the level obtained in the control. At 100 µmol/l of oleic acid, a concentration that increased amylase and lipase secretion without inducing LDH release (see fig 4B), the increase in [Ca2+]cyt was very small and a CCK signal similar to that of the control was elicited (see fig 5A,B). However, the second phase of the increase in [Ca2+]cyt was recovered and the curve resembled that with CMs alone when oleic acid was added together with CMs in the presence of orlistat. This indicates that the second delayed phase of increase in [Ca2+]cyt was due to the generation of FFAs in the presence of CMs alone. A delayed increase in fatty acid generation from CMs (1 mg/ml) at the expense of a decrease in triglyceride concentration after 20 min can be also seen from fig 3A,D. Whereas the first phase increase in [Ca2+]cyt was reversible by washing cells with buffer during a 30 min superfusion period, the second phase was irreversible (data not shown) and indicates damage to the cells.
Lipase-induced Ca2+ signalling
The release of amylase induced by active as well as inactive lipase without cell damage (fig 4C) led us to hypothesise that the lipase protein itself has a signalling effect on pancreatic acinar cells. Since regulated pancreatic enzyme secretion requires mainly cellular Ca2+ signalling, we tested the effects of active and inactive lipases on cytosolic Ca2+ concentrations in pancreatic acinar cells. Addition of active lipase (500 U/ml) or of inactive lipase (170 µg/ml corresponding to 500 U/ml of active lipase) to cells did not show any effect on [Ca2+]cyt different from the control (fig 6A). However, the global Ca2+ signal induced by CCK (100 pmol/l) was converted to Ca2+ oscillations in the presence of active lipase (500 U/ml), lipase inhibited by orlistat or the inactive lipase HPLRP1. This indicates a direct effect of the lipase protein on [Ca2+]cyt irrespective of FFA generation by enzymatic action of lipase (figs 5B and 6A).

Ca2+ oscillations were also seen at very low CCK concentrations (10−12 mol/l) with or without lipase (fig 6B). However, addition of inactive lipase shifted the CCK-induced oscillatory increase in [Ca2+]cyt from a CCK concentration of 10−11 mol/l to CCK concentrations as high as 10−9 mol/l. Since lipase seems to act like an agonist a specific receptor of the enzyme at the cell surface would be required. Since heat-inactivated (and probably unfolded or partly unfolded) lipase had a similar effect to the well-folded but inactive lipase related protein (HPLRP1) and lipase mutant (HPL(S152G)) it is possible that a short amino acid sequence in the protein is the true ligand. Recently, porcine pancreatic phospholipase A2 (PLA2) was found to stimulate secretin release independently of its enzymatic activity.19 Furthermore, two secretin-releasing peptides (SRP 1 and SRP 2) were isolated from canine pancreatic juice.20 The sequence of SRP 1 was identical to the first 25 N-terminal amino acids of PLA2, and SRP 2 showed 71% homology to the first 21 amino acids of PLA2.20 Similar to our results, these experiments demonstrate that a protein may act as signal transducer irrespective of its enzymatic activity.
Lipase-induced generation of cGMP
Since both active and inactive lipase act like an agonist on pancreatic acinar cells to induce amylase secretion (fig 4C) and Ca2+ oscillations (fig 6A,B), we tested whether the transduction of these molecules also signals with generation of a second messenger in the cell. Indeed, cGMP production was increased by inactive lipase (HPL(S152G), 170 µg/ml) in a similar manner to the secretagogue CCK (fig 7A). The level of another second messenger, cAMP, which is produced in the presence of the pancreatic secretagogue secretin was not changed by the presence of inactive lipase (fig 7A). Similarily, active pancreatic lipase also increased the cellular cGMP level (fig 7B); this effect was not inhibited by orlistat, an inhibitor of lipase enzymatic activity. However, an unexpected finding was stimulation of cGMP production by orlistat. We have no explanation for this finding at present. We can only speculate that orlistat binds to the same binding site as lipase in pancreatic acinar cells. Also, other inhibitors such as atropine (an antagonist of the acetylcholine receptor) or proglumide (an antagonist of CCK receptors) did not inhibit the effect of lipase (see fig 7B). The agonist action of pancreatic lipase seems to recruit a different intracellular pathway which leads to enzyme release without any significant elevation in cytosolic [Ca2+]. We have previously provided evidence that cytosolic ADP ribosyl cyclase is activated by CCK, most likely mediated by cGMP.21 The same enzyme can also produce nicotinamide adenine dinucleotide phosphate (NAADP). Ca2+ spikes in the apical pole of pancreatic acinar cells are elicited by low concentrations of CCK via different Ca2+ release channels.22 Ca2+ oscillations are triggered by NAADP via its receptor at low CCK concentrations and amplified by cADPR via the ryanodine receptor. CCK-induced Ca2+ spikes are turned to sustained global Ca2+ increase at higher CCK concentrations when the third messenger inositol-4,5-bisphosphate (IP3) comes into play.21 The NAADP receptor becomes desensitised even at NAADP concentrations that are below the threshold of Ca2+ release.23–25 At high CCK concentrations (10−9 mol/l) at which the concerted action of all three messengers leads to a large, sustained global Ca2+ elevation, desensitisation of the receptor of one of these messengers (NAADP) would return CCK-induced Ca2+ response to oscillation.22 26–28. It is tempting to hypothesise that lipase leads to cGMP-mediated activation of ADP ribosyl cyclase with generation of cADP ribose and/or of NAADP and consequently to the abolishment of CCK-induced, NAADP-mediated Ca2+ release by desensitisation of NAADP receptors. This would return CCK-mediated global Ca2+ elevations to Ca2+ oscillations as described here (see fig 6 A,B). Further studies are required to validate the interpretation of the phenomenon observed in our experiments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
We conclude from our data that FFAs are effective stimulators of enzyme secretion from pancreatic acinar cells and cause cell damage at high concentrations. High concentrations per se as found in patients with HTG and in LPL-deficient mice, however, cannot explain cell damage that could initiate pancreatitis. Although in patients with high HTG levels and with pancreatitis, high plasma concentrations of total FFAs are frequently present (>1 mmol/l; see Sztefko and Panek17). They are bound to plasma proteins and are not expected to pass blood capillaries and reach pancreatic acinar cells. High concentrations of CMs and fatty acids in the serum therefore should not, per se, be the reason for the development of pancreatitis either in patients or in LPL-deficient mice with high triglyceride levels. In fact, LPL-deficient mice only developed pancreatitis after long-term stimulation with caerulein (see figs 1 and 2). It appears that pancreatic cells are more susceptible to damage by an additional stimulus or injury in the presence of high triglyceride plasma levels surrounding pancreatic cells. Damage to cells as a result of different reasons and other factors, such as infection, alcohol abuse or genetic predisposition, are all risk factors for initiation and manifestation of pancreatitis in patients with high plasma concentrations of triglycerides. Once cells are damaged, high concentrations of pancreatic lipase exceeding those normally present in the blood plasma can reach the interstitium. In patients with high triglyceride plasma levels pancreatic cell damage could be aggravated by continuous breakdown of chylomicrons, generation of fatty acids which leads to further cell damage and to increase in cytosolic Ca2+ levels. Progressive disruption of acinar cell calcium signalling is an early feature of caerulein-induced pancreatitis in mice.29 Pancreatic lipase seems to have a dual function. In addition to its triglyceride hydrolysing activity lipase reaching the interstitial cell side can also directly act as an agonist at the acinar cell membrane. In this agonist function it would even protect pancreatic cells by converting global Ca2+ signals to Ca2+ oscillations which prevents Ca2+ overload.
Acknowledgments
IS thanks her colleagues Professor Dr J Rettig and Professor Dr M Hoth, Institute of Physiology, Saarland University; and Professor Dr V Flockerzi, Institute of Pharmacology, Saarland University, for continuous support of her studies. She also thanks G Moerschbaecher for long-lasting secretarial assistance; and H Konzmann, P Hammes and S Plant for excellent technical assistance.
REFERENCES
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Supplementary methods, figures and a table are published online only at http://gut.bmj.com/content/vol58/issue3
-
YW, LS and FY contributed equally to this work.
-
Funding: The authors thank the Deutsche Forschungsgemeinschaft (DFG) for support of LS, FY and IS in the SFB 390, project 5A. They also thank the Major National Basic Research Program of the People’s Republic of China (G2006CD5o3801), Program for the Changjiang Scholars Innovative Research Team in University (PCSIRT).
-
Competing interests: None.
-
Ethics approval: The Principles of Laboratory Animal Care (NIH publication no. 85–23, revised 1996) was followed and the experimental protocol was approved by the Animal Care Committee, Peking University Health Science Center.