Article Text

Abstract
Background and Aims: In addition to its crucial role in dampening tissue-damaging immune responses in the gut, transforming growth factor β (TGFβ) is a potent profibrogenic agent inducing collagen synthesis and regulating the balance between matrix-degrading matrix metalloproteinases (MMPs) and their inhibitors (TIMPs). TGFβ signalling was investigated by analysis of Smad proteins and MMPs/TIMPs in the mucosa overlying strictures in patients with Crohn’s disease (CD).
Methods: Specimens were collected from macroscopically normal mucosa overlying strictured and non-strictured gut of patients with fibrostenosing CD. Isolated myofibroblasts were cultured with anti-TGFβ blocking antibody or TGFβ1. TGFβ transcripts were analysed by quantitative reverse transcription-PCR (RT-PCR). Smad proteins and MMPs were determined by immunoblotting. MMP-12 activity was measured by a real-time MMP-12 activity assay. An in vitro wound-healing scratch assay was used to assess myofibroblast migration.
Results: TGFβ transcripts, phosphorylated Smad2–Smad3 (pSmad2–3) and TIMP-1 proteins were higher in mucosa overlying strictures than in mucosa overlying non-strictured areas. In contrast, mucosa overlying strictured gut had lower expression of Smad7, MMP-12 and MMP-3. Myofibroblasts from mucosa overlying strictured gut showed higher TGFβ transcripts, a greater pSmad2–3 response to TGFβ, increased TIMP-1, lower Smad7, increased collagen production and reduced migration ability compared with myofibroblasts from mucosa overlying non-strictured gut. TGFβ blockade increased myofibroblast MMP-12 production and migration, more obviously in myofibroblasts isolated from mucosa overlying non-strictured compared with strictured gut.
Conclusions: Changes in TGF-β signalling and MMP production were identified in the mucosa overlying strictures in CD which may give a window into the process of fibrosis.
Statistics from Altmetric.com
Intestinal fibrosis and stricture formation are often part of the natural course of Crohn’s disease (CD), frequently requiring surgical resection.1 In this situation, excessive synthesis and deposition of extracellular matrix (ECM) components by intestinal myofibroblasts, as well as inhibition of ECM degradation due to an imbalance between matrix metalloproteinases (MMPs) and their inhibitors (TIMPs), are thought to be involved in the fibrogenic process.2 3 In a murine model of chronic inflammation, fibrosis is associated with an increase in TIMP-1 which results in inhibition of MMP-mediated ECM degradation.4 Increased TIMP-1 has also been shown in collagenous colitis and colonic diverticular disease,5 6 again suggesting that fibrosis may be due to inhibition of ECM degradation by MMPs as well as increased ECM production per se.
Transforming growth factor β (TGFβ) is the protypic profibrogenic cytokine involved in fibrosis in many organ systems, including intestinal strictures in CD.7 In particular, the TGFβ1 isoform has been specifically implicated in fibrosis through its ability to promote ECM synthesis and fibroblast contraction.8 Both TGFβ and its receptors are overexpressed in the intestine of patients with CD,9 and their binding induces the activation of the Smad transcriptional proteins. TGFβ receptor I kinase directly phophorylates Smad2 and Smad3 that then bind to the common mediator Smad4 and translocate to the nucleus to regulate gene transcription. Inhibitory Smad proteins (Smad6 and Smad7) act by competing for the TGFβ receptor I kinase and inibiting Smad phosphorylation.10 11
Growth factors also promote wound healing by stimulating ECM synthesis, in part by modulating the balance between MMPs and TIMPs.1 12 CD myofibroblasts isolated from strictured bowel constitutively overexpress collagen type III and fibronectin, and the production of ECM components and that of collagen type I is increased by TGFβ.13 The latter may facilitate the fibrogenic process by stimulating TIMP-1 production or inhibiting MMP expression.14–16 Moreover, TGFβ1 has been shown to promote enhanced myofibroblast contractile activity, which is a key event predisposing to scar contraction and stricture formation.2 17
In contrast to the progress in understanding the inflammatory aspects of CD, progress in understanding fibrosis has been slow. This is largely because fibrosis is predominantly of the outer muscle layers which cannot be sampled during endoscopy. Furthermore, tissue taken from resected bowel often represents end-stage disease where early pathogenic events have been obscured. In this study we have therefore decided to focus on the mucosa overlying strictured bowel, to determine if it contains any signatures of altered TGFβ signalling and MMP production which could be informative of events in the deeper layers of the gut. We focused on MMP-3 (stromelysin-1) and MMP-12 (macrophage metalloelastase) as these two enzymes have been shown to play a pathogenic role in CD.12 18 MMP-3, which cleaves proteoglycans, collagens (type II, IX and XI), gelatin, laminin-1 and fibronectin,19 20 is involved in the development of mucosal ulceration and fistulas in CD, where it is released by lamina propria Th1 cytokine-activated myofibroblasts or immunoglobulin G (IgG) plasma cells.21–24 MMP-12 is a macrophage-specific enzyme, which degrades different substrates, including elastin, laminin, type IV collagen, fibronectin and casein.
Our null hypothesis is that we would find no difference in TGFβ signalling between normal mucosa from control individuals and normal mucosa overlying strictures. Based on enhanced TGFβ signalling, less MMP activity and increased collagen production by myofibroblasts from mucosa overlying strictured bowel, we can reject our null hypothesis.
PATIENTS AND METHODS
Patients
Endoscopic biopsies or surgical specimens were taken from macroscopically uninflamed ileal or colonic mucosa of strictured and non-strictured areas of 25 patients with fibrostenosing CD (table 1). They had suspended steroid treatment at least 3 months earlier, and none of them had ever been treated with ciclosporin, methotrexate, azathioprine or infliximab. Diagnosis of CD was ascertained according to the usual clinical criteria,25 and strictured areas were identified by conventional or CT enteroclysis.26 Mucosal samples were also collected from the ileum or colon of 20 subjects who had functional diarrhoea at the end of their diagnostic investigation (mean age 34.8 years, range 22–68), and from macroscopically and microscopically unaffected colonic areas of 14 patients undergoing colectomy for colon cancer (mean age 37.6 years, range 25–68), and from macroscopically and microscopically inflamed areas of 18 patients affected by non-fibrostenosing inflammatory CD (table 1). Ten patients affected by diverticular disease (mean age 58.6 years, range 45–71) were also studied as a disease-control group. The median histological score, graded from 0 to 3 as previously described,27 did not differ significantly in strictured (0, range 0–1) and non-strictured mucosa (0, range 0–1) of patients with fibrostenosing CD and control mucosa (0, range 0–1), while it was significantly higher (p<0.001) in inflamed mucosa of patients with non-fibrostenosing inflammatory CD (2.0, range 1–3). Some of the mucosal samples were used to isolate myofibroblasts and some were homogenised and used for immunoblotting and quantitative reverse transcription-PCR (RT-PCR). Each patient who took part in the study was recruited after appropriate local Ethics Committee approval, and informed consent was obtained in all cases.
Cell isolation and culture
Mucosal myofibroblasts were isolated as previously described.16 Briefly, the epithelial layer was removed by 1 mM EDTA (Sigma-Aldrich, Poole, UK) for two 30 min periods at 37°C. After EDTA treatment, mucosal samples were denuded of epithelial cells, and were subsequently cultured at 37°C in a humidified CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 20% fetal calf serum (FCS), 1% non-essential amino acids (Invitrogen, Paisley, UK), 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamycin and 1 μg/ml amphotericin (Sigma-Aldrich). During culture, numerous cells appeared both in suspension and adherent to the culture dish. The cells in suspension were removed after every 24–72 h culture period, and the denuded mucosal tissue was maintained in culture for up to 6 weeks. Established colonies of myofibroblasts were seeded into 25 cm2 culture flasks and cultured in DMEM supplemented with 20% FCS and antibiotics. At confluence, the cells were passaged using trypsin-EDTA in a 1:2 to 1:3 split ratio. Cells were grown up to at least passage 4 (P4) before there were enough to use in stimulation experiments, and were characterised by immunocytochemical staining, as previously described.16 All the experiments were performed with cells at low passage (P4–P6), with the exception of those conducted on high passage (P16–P20) control and non-strictured cells to assess the influence of senescence on myofibroblast behaviour.
Cell stimulation
Subconfluent monolayers of myofibroblasts seeded in 12-well plates at 3×105 cells per well were starved for 24 h before incubation for 24 h with 10 ng/ml recombinant human TGFβ1 (R&D Systems, Abingdon, UK), 20 μg/ml pan-neutralising anti-TGF-β monoclonal antibody (R&D Systems) or its isotype control (rabbit IgG, Sigma-Aldrich).
Wound-healing scratch assay
Myofibroblast migration was assessed according to the method of Rodriguez et al28 modified by us.16 Briefly, cells (2×105) were seeded into Nunc cell culture dishes (Nalge Nunc International, Rochester, New York, USA) with 2 mm grids, size 35× 10 mm, in 2 ml of DMEM supplemented with 20% FCS and antibiotics. The cells were maintained at 37°C and 5% CO2 until confluent. Once confluent, each dish of monolayer cells was given a mechanical wound by scoring with a 200 μl pipette tip, parallel to the grid bars along the central grid line. Wound placement was checked with an Olympus inverted CK2 microscope (Olympus UK, London, UK). The medium was then removed, and the cells were washed five times with HL-1 serum-free medium (Cambrex Bio Science, Nottingham, UK) supplemented with antibiotics, and then replaced with 1.5 ml of HL-1 medium with the following treatments: TGFβ1 (10 ng/ml), anti-TGFβ antibody (20 μg/ml) or its isotype control (rabbit IgG). Photographs of the cells in each grid along the induced wound were taken at 0, 2, 4, 8, 16 and 24 h, using a digital camera (Olympus Camedia 34–40 zoom, ×20 magnification) attached to a light microscope. The computer program “Image J” was used to measure the area of initial damage (images taken at time 0) and of the remaining damage at subsequent time points. Each grid image was observed separately; two points per grid at the same position at every time point were measured using imaging software at the same magnification. The percentage of wound repair was then calculated.
Western blotting
Western blotting was performed according to a modified method described previously.29 In brief, cells or tissue samples were lysed in ice-cold lysate buffer (10 mM EDTA, 50 mM pH 7.4 Tris–HCl, 150 mM sodium chloride, 1% Triton X-100, 2 mM phenylmethylsulfonyl fluoride, 2 mM sodium orthovanadate, 10 mg/ml leupeptin and 2 mg/ml aprotinin) and the amount of protein was determined by the Bio-Rad Protein assay (Bio-Rad Laboratories, Hemel Hempstead, UK). A 10 μg aliquot of protein or 15 μl of cell culture supernatants was loaded in each lane and subjected to 10% sodium dodecylsulfate–polyacrylamide get electophoresis (SDS–PAGE) under reducing conditions. After electrophoresis, protein was transferred to nitrocellulose (Bio-Rad Laboratories). The following primary antibodies were used: sheep anti-MMP-3 (1:500 dilution, The Binding Site, Birmingham, UK), mouse anti-MMP-12 (1:200 dilution, R&D Systems), mouse anti-TIMP-1 (1 μg/ml, Oncogene Research, Nottingham, UK), goat anti-pSmad2–3 (1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, California, USA) and goat anti-Smad7 (1:500 dilution, Santa Cruz Biotechnology). Appropriate antibodies conjugated to horseradish peroxidase (DAKO, High Wycombe, UK) were used as secondary antibodies, and the reaction was developed with the ECL plus kit (Amersham Biosciences, Little Chalfont, UK). When required, blots were stripped and analysed for β-actin, as an internal loading control, using a rabbit anti-β-actin antibody (1:5000 dilution, AbCam, Cambridge, UK), or for Smad3, a rabbit anti-Smad-3 antibody (1:500 dilution, Santa Cruz Biotechnology). Bands were quantified by scanning densitometry using an LKB Ultrascan XL Laser Densitometer (Kodak, Hemel Hempstead, UK).
RNA extraction and analysis of mRNA expression by quantitative RT-PCR
RNA was extracted from biopsies using the following method. Frozen tissue was added to tubes containing lysing matrix D beads (Bio101) plus 600 μl of lysis buffer from the RNA microprep kit (Qiagen, Crawley, UK) and the tissue was disaggregated using a Ribolyzer (Hybaid Heidelberg, Germany) for 40 s. Lysates were placed on ice then RNA was extracted using the Qiagen RNA microprep kit according to the manufacturer’s instructions. cDNAs were synthesised with the Improm-II RT system (Promega, Southampton, UK) using random hexamers and 1 μg of RNA in a final volume of 20 μl. Reverse transcription reactions were performed using the Improm-II reverse transcriptase enzyme. A reverse transcription reaction without reverse transcriptase enzyme was performed for each tissue type as a negative control for quantitative PCR. TGFβ primer and probe sets were validated for use with the ΔΔCT method of quantification (see “User bulletin 2 ABI7700 sequence detection system” for details). The probe was labelled with a 5′-reporter dye FAM (6-carboxy-fluorescein) and the 3′-quencher dye TAMRA (6-carboxy-N,N,N’,N′-tetramethyl-rhodamine). Primers against 18S rRNA and a FAM-labelled probe were used as a normalising control. Reverse transcription reactions were diluted 1 in 10 in distilled H2O, and 5 μl of template was added to 6.5 μl of 2× master mix (Eurogentech, Seraing, Belgium) containing 1.2 μM forward and reverse primers and 0.248 μM of probe in a total volume of 12.5 μl. The PCR protocol was as follows: 50°C, 2 min, 95°C, 10 min, followed by 40 cycles of denaturation 95°C, 15 s, and annealing/extension at 60°C, 1 min. Thermocycling and real-time detection of PCR products were performed on an IcyclerIQ sequence detection system (Bio-Rad Laboratories) and, following completion of the PCR, the thresholds for fluorescence emission baseline were set just above background levels. Expression levels were normalised with 18S and values were calculated using the ΔΔCT method and expressed relative to one of the specimens that was assigned the value 1. Error bars represent 1 standard deviation, and p values, where shown, were calculated using the Mann–Whitney rank sum test.
Real-time MMP-12 activity assay
The activity of MMP-12 in the culture supernatants was detected and measured using an EnzoLyte MMP-12 assay kit (AnaSpec Corporate, San Jose, California, USA) according to the manufacturer’s instructions. A 25 μl aliquot of culture supernatant was incubated with or without p-aminophenyl mercuric acetate (APMA, final concentration 1 mM) for 15 min at 37°C immediately before the measurement. The supernatant was then incubated with an equal volume of MMP-12 substrate solution in a PCR plate (ABgene, Epsom, Surrey, UK). The reagents were mixed by shaking the plate gently for 30 s. The fluorescence intensity at excitation/emission = 490 nm/520 nm was measured and recorded in relative fluorescence units (RFU) every 5 min for 100 min using an iQ5 real-time PCR thermal cycler (Bio-Rad Laboratories). The fluorescence substrate used in this assay was 5-FAM/QXL520 fluorescence resonance energy transfer peptide. In the intact fluorescence resonance energy transfer peptide, the fluorescence of 5-FAM is quenched by QXL520. Upon cleavage into two separate fragments by MMP-12, the fluorescence of 5-FAM is recovered and can be monitored. This assay is able to detect as little as 10 pg/μl of MMP-12. It has high specificity for MMP-12 and minimal cross-reactions with other MMPs. Tests were also carried out with an anti-MMP-3 antibody (R&D Systems) and an anti-TIMP-1 antibody (Calbiochem, Nottingham, UK), and it was shown that there was no cross-reaction with MMP-3 and TIMP-1.
Flow cytometry
Apoptosis was quantified using APC (allophycocyanin)–Annexin V (BD Biosciences, Oxford, UK) and propidium iodide by flow cytometry with a FACSCalibur Flow Cytometer (BD Biosciences). The human Jurkat T cell line, stimulated in anti-CD3-coated 96-well plates (BD Biosciences) with an anti-CD28 antibody (0.5 μg/ml, eBioscience, San Diego, California, USA) and then incubated for 24 h with 10 μg/ml infliximab, was used as positive control.
Collagen assay
Total soluble forms of collagen were measured in myofibroblast supernatants using the Sircol Collagen Assay Kit (Biocolor, Belfast, UK) according to the manufacturer’s instructions. The Sircol dye reagent has been formulated to bind specifically to the (Gly-X-Y)n helical structure found in collagen types I–XIV. The collagen content in each sample of cell supernatant was obtained as an average of three readings.
Cell senescence detection
Myofibroblast senescence was assessed using the Senescence Detection Kit (BioVision, Mountain View, California, USA), which is designed to detect senescence-associated expression of β-galactosidase activity in cultured cells histochemically, according to the manufacturer’s instructions.
In situ collagen and α-smooth muscle cell actin (SMA) staining
Collagen was analysed on paraffin sections (5 μm thick) with Masson’s trichrome. Immunohistochemistry was performed on serial sections according to established methods by using an anti-SMA antibody (clone 1A4; DAKO). The sections were examined using conventional light microscopy in a blinded manner by the same expert observer.
Statistical analysis
Data were analysed in the GraphPad Prism statistical PC program (GraphPad Software, San Diego, California) using the paired t test and the Mann–Whitney U test. A level of p<0.05 was considered statistically significant.
RESULTS
In our analysis we compared uninflamed CD mucosa overlying strictured gut, uninflamed CD mucosa overlying non-strictured gut, inflamed mucosa from patients with non-fibrostenosing inflammatory CD, and normal mucosa from patients with non-inflammatory bowel disease and patients with diverticular disease. The last three groups were included as controls, for example to be sure that Smad7 was high in inflamed gut, as previously reported.30
Mucosal TGFβ transcripts and Smad proteins
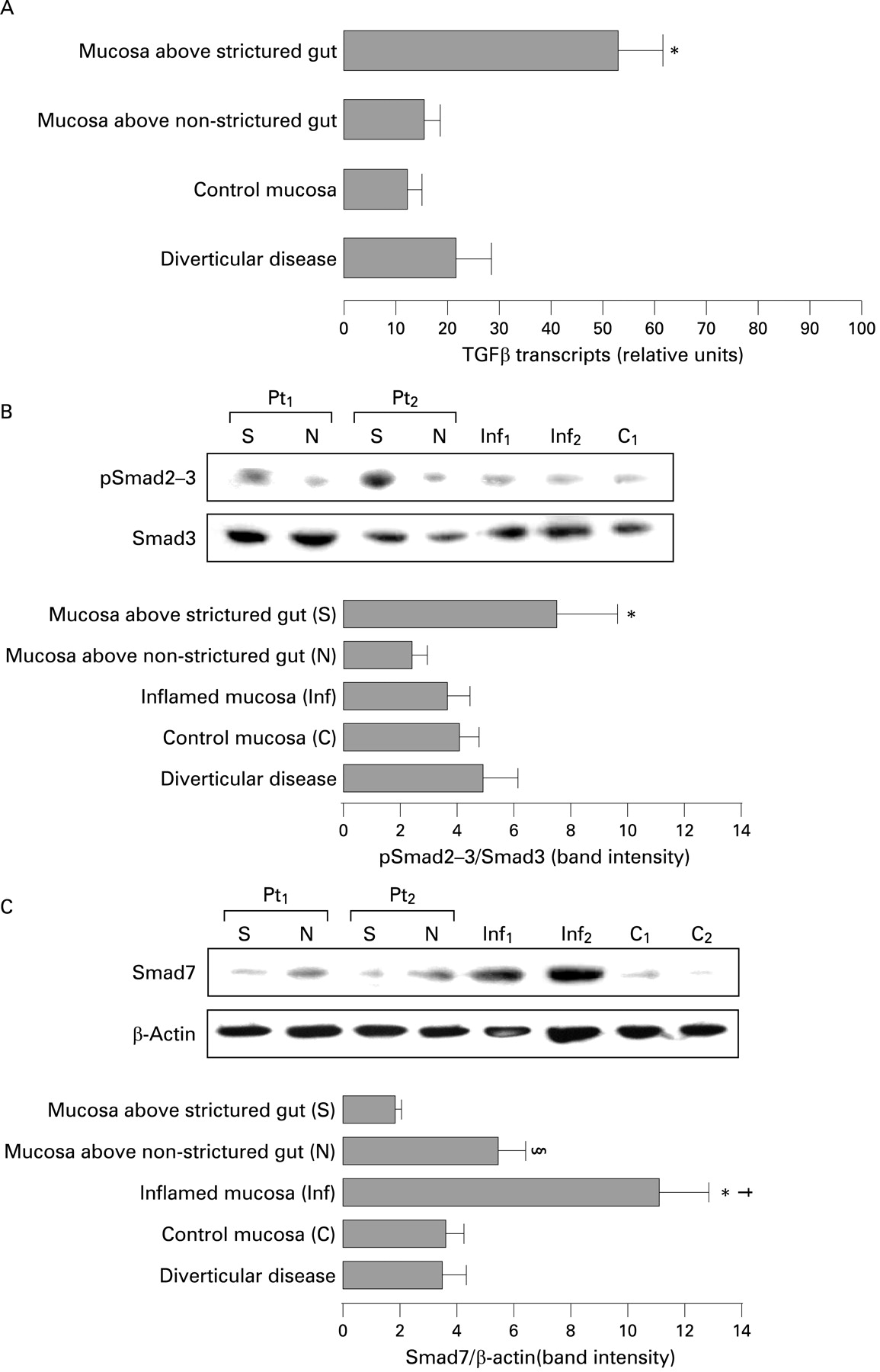
We first investigated TGFβ transcripts in mucosa overlying strictured and non-strictured gut of 20 patients with fibrostenosing CD by quantitative RT-PCR. We found a significantly (p<0.001) higher number of TGFβ transcripts in mucosa overlying CD strictures than in mucosa overlying non-strictured areas and normal mucosa of 18 control subjects (fig 1A). Secondly, as phosphorylation of Smad2–3 proteins is a key step in activating TGFβ signalling,10 11 we determined by immunoblotting the mucosal level of pSmad2–3. Significantly (p<0.05) higher expression of pSmad2–3 was found in mucosa overlying strictured gut compared with mucosa overlying non-strictured areas in patients with fibrostenosing CD (fig 1B). Thirdly, as Smad7 is a negative regulator of TGFβ signalling, we also determined mucosal Smad7 expression (fig 1C). A significantly (p<0.05) lower expression of Smad7 was found in mucosa overlying strictured gut in comparison with mucosa overlying non-strictured areas. As expected,30 Smad7 expression was significantly (p<0.01) higher in inflamed mucosa of 18 patients with non-fibrostenosing inflammatory CD in comparison with mucosa overlying non-strictured gut and control mucosa. TGFβ transcripts, pSmad2–3 and Smad7 did not differ significantly in the 10 patients with diverticular disease in comparison with control subjects.

(A) Detection of transforming growth factor β (TGFβ) transcripts by quantitative reverse transcription-PCR (RT-PCR) in uninflamed mucosa overlying strictures and non-strictured areas from 20 patients with fibrostenosing Crohn’s disease (CD), and in normal mucosa from 18 control subjects and 10 patients with diverticular disease. Results are means (SD) (*p<0.001 vs CD mucosa overlying non-strictured areas, normal mucosa and diverticular disease mucosa). (B) Detection of phosphorylated Smad2 and Smad3 proteins (pSmad2–3) by immunoblotting in uninflamed mucosa overlying strictures (S) and non-strictured areas (N) of two patients with fibrostenosing CD (Pt1 and Pt2), inflamed mucosa of two patients with non-fibrostenosing inflammatory CD (Inf1 and Inf2) and normal mucosa from a control subject (C1). Blots were stripped and analysed for Smad3 as an internal loading control. Each example is representative of experiments performed in 20 patients with fibrostenosing CD, 18 patients with non-fibrostenosing inflammatory CD and 18 control subjects. The lower panel shows densitometry of western blots. Results are means (SD) (*p<0.05 vs CD mucosa overlying non-strictured areas, CD inflamed mucosa, normal mucosa and mucosa from 10 patients with diverticular disease). (C) Detection of Smad7 by immunoblotting in uninflamed mucosa overlying strictures (S) and non-strictured areas (N) of two patients with fibrostenosing CD (Pt1 and Pt2), inflamed mucosa of two patients with non-fibrostenosing inflammatory CD (Inf1 and Inf2) and normal mucosa from two control subjects (C1 and C2). Blots were stripped and analysed for β-actin as an internal loading control. Each example is representative of experiments performed in 20 patients with fibrostenosing CD, 18 patients with non-fibrostenosing inflammatory CD and 18 control subjects. The lower panel shows densitometry of western blots. Results are the mean (SD). (§p<0.05 vs CD mucosa overlying strictures; *p<0.01 vs CD mucosa overlying strictures, control mucosa and mucosa from 10 patients with diverticular disease; †p<0.05 vs CD mucosa overlying non-strictured areas)
Mucosal MMP-12, MMP-3 and TIMP-1 proteins
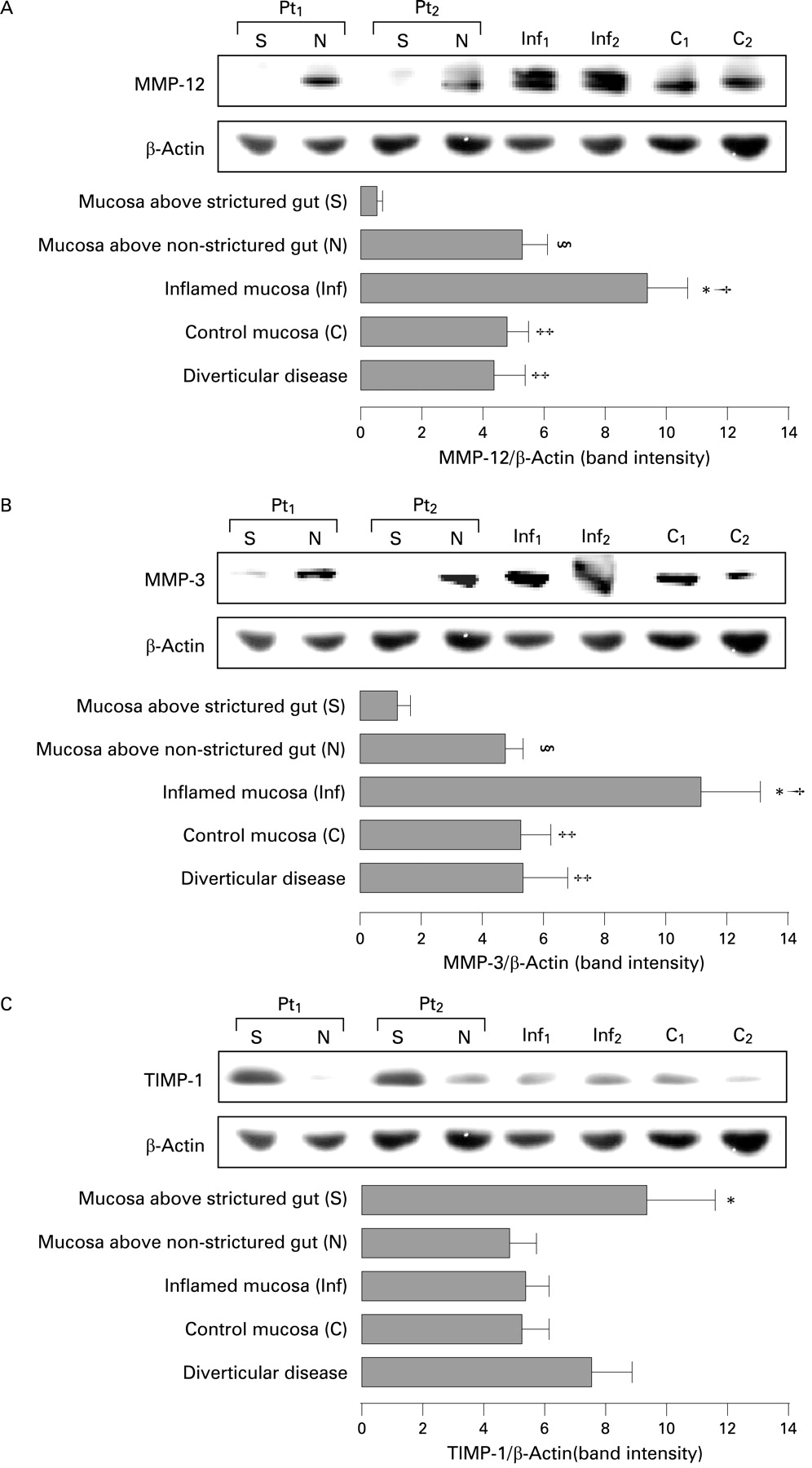
MMP-12 (macrophage metalloelastase), MMP-3 (stromelysin-1) and TIMP-1 expression was determined by immunoblotting in mucosa overlying strictures and non-strictured areas of 20 patients with fibrostenosing CD. A significantly (p<0.05) lower expression of MMP-12 was found in mucosa overlying strictures in comparison with mucosa overlying non-strictured areas (fig 2A). MMP-12 expression was significantly higher in inflamed mucosa of 18 patients with non-fibrostenosing inflammatory CD in comparison with normal mucosa from 18 control subjects (p<0.05), CD mucosa overlying non-strictured (p<0.05) or strictured gut (p<0.01) and mucosa from 10 patients with diverticular disease (p<0.05). For MMP-3, significantly (p<0.05) lower expression was found in mucosa overlying strictures compared with mucosa overlying non-strictured areas (fig 2B). MMP-3 expression was significantly higher in CD inflamed mucosa in comparison with control mucosa (p<0.05), CD mucosa overlying non-strictured (p<0.05) or strictured gut (p<0.01) and diverticular disease mucosa. We also determined TIMP-1 expression that was significantly (p<0.05) higher in mucosa overlying CD strictures than mucosa overlying non-strictured areas (fig 2C). Moreover, TIMP-1 did not differ significantly among CD non-strictured areas, CD inflamed areas and normal mucosa. Patients with diverticular disease (n = 10) showed comparable levels of mucosal MMP-3 and MMP-12 with those of control subjects, while TIMP-1 was increased, though not significantly, in comparison with normal mucosa.

Detection of matrix metalloproteinase-12 (MMP-12) (A), MMP-3 (B) and tissue inhibitor of metalloproteinase 1 (TIMP-1) (C) in uninflamed mucosa overlying strictures (S) and non-strictured areas (N) of two patients with fibrostenosing Crohn’s disease (CD) (Pt1 and Pt2), inflamed mucosa of two patients with non-fibrostenosing inflammatory CD (Inf1 and Inf2) and normal mucosa from two control subjects (C1 and C2). Blots were stripped and analysed for β-actin as an internal loading control. Each example is representative of experiments performed in 20 patients with fibrostenosing CD, 18 patients with non-fibrostenosing inflammatory CD and 18 control subjects. Histograms show densitometry of western blots. Results are means (SD). (A: §p<0.05 vs CD mucosa overlying strictures; *p<0.01 vs CD mucosa overlying strictures; †p<0.05 vs CD mucosa overlying non-strictured areas, control mucosa and mucosa from 10 patients with diverticular disease; ‡p<0.05 vs CD mucosa overlying strictures. B: §p<0.05 vs CD mucosa overlying strictures; *p<0.01 vs CD mucosa overlying strictures; †p<0.05 vs CD mucosa overlying non-strictured areas, control mucosa and mucosa from 10 patients with diverticular disease; ‡p<0.05 vs CD mucosa overlying strictures. C: *p<0.05 vs CD mucosa overlying non-strictured areas, CD inflamed mucosa, normal mucosa and mucosa from 10 patients with diverticular disease.)
TGFβ transcripts, Smad proteins and collagen production by myofibroblasts
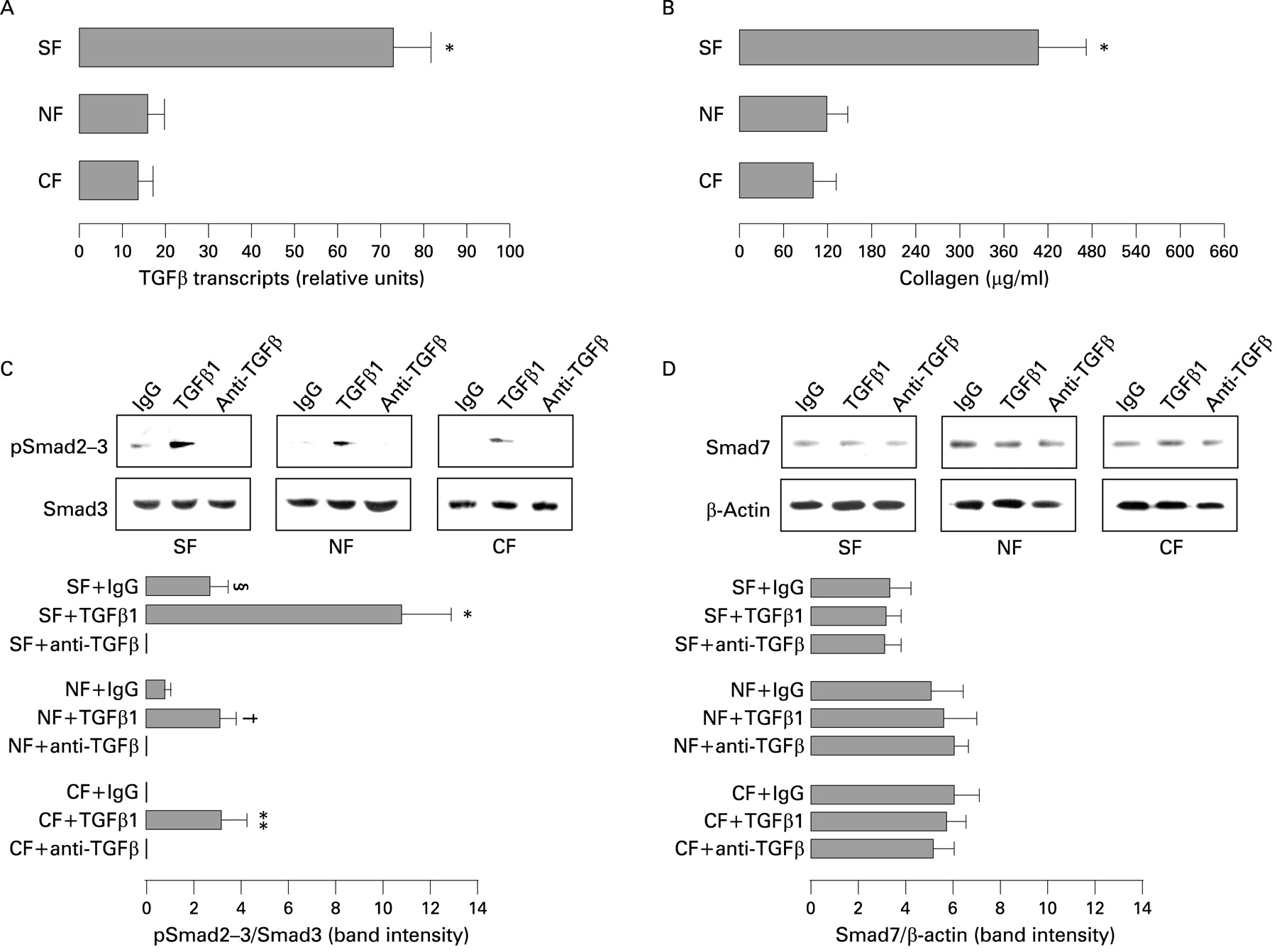
A significantly (p<0.001) higher number of TGFβ transcripts was found in myofibroblasts from mucosa overlying strictures in comparison with myofibroblasts from mucosa overlying non-strictured areas of 18 patients with fibrostenosing CD and normal myofibroblasts from 16 control subjects (fig 3A). Moreover, myofibroblasts from mucosa overlying strictures produced significantly (p<0.001) higher amounts of collagen (mean (SD) 402 (79) μg/ml) compared with myofibroblasts from mucosa overlying non-strictured gut (116 (30) μg/ml) and control myofibroblasts (98 (29) μg/ml) (fig 3B). No significant difference was found between non-strictured myofibroblasts and control myofibroblasts in terms of TGFβ transcripts and collagen production. pSmad2–3 expression was significantly (p<0.01) increased when myofibroblasts from mucosa overlying strictures were stimulated with TGFβ1 in comparison with IgG-treated cells. A significant (p<0.05) increase was also observed in TGFβ1-stimulated myofibroblasts from mucosa overlying non-strictured gut. TGFβ blockade significantly (p<0.05) inhibited the expression of pSmad2–3 in myofibroblasts from mucosa overlying strictures in comparison with IgG-treated cells (fig 3C). Additionally, control myofibroblasts showed lower levels of pSmad2–3 expression compared with the former two groups when treated with control IgG. No difference was found in the expression of Smad7 in myofibroblasts from any group of patients, regardless of different stimuli (IgG, anti-TGFβ antibody or TGFβ1) (fig 3D). In keeping with the in vitro experiments, we observed in vivo an increased expression of collagen in the mucosa overlying strictures in comparison with the mucosa overlying non-strictured areas (fig 4) of three patients with fibrostenosing CD. Additionally, SMA-positive cells appeared morphologically different in the mucosa overlying strictures in comparison with non-strictured areas, showing a round shape and numerous projections which lengthen in the lamina propria in strictured mucosa, while they were fusiform and mostly located in the pericryptal region in non-strictured mucosa (fig 4).

(A) Detection of transforming growth factor β (TGFβ) transcripts by quantitative reverse transcription-PCR (RT-PCR) in myofibroblasts isolated from uninflamed mucosa overlying strictures (SF) and non-strictured areas (NF) from 18 patients with fibrostenosing Crohn’s disease (CD), and normal myofibroblasts from 16 control subjects (CF). Results are means (SD) (*p<0.001 vs NF and CF). (B) Soluble collagen was measured in the supernatants of SF and NF from 18 patients with fibrostenosing CD, and in the supernatants of CF from 16 control subjects. Results are means (SD) (*p<0.001 vs NF and CF). (C and D) Detection of phosphorylated Smad2 and Smad3 (pSmad2–3) (C) and Smad7 (D) in SF, NF and CF cultured with TGFβ1, anti-TGFβ antibody or its isotype-matched control (rabbit immunoglobulin G (IgG)). pSmad2–3 and Smad7 blots were stripped and respectively analysed for Smad3 and β-actin as an internal loading control. Each example is representative of experiments performed in 18 patients with fibrostenosing CD and 16 control subjects. Histograms show densitometry of western blots. Results are means (SD). (C: §p<0.05 vs anti-TGFβ-treated SF; *p<0.01 vs IgG-treated SF; †p<0.05 vs IgG-treated NF; **p<0.05 vs IgG-treated CF.)

Histochemical detection of collagen by Masson’s trichrome and immunohistochemical detection of α-smooth muscle cell actin (SMA) in the mucosa overlying strictured and non-strictured areas in a patient with fibrostenosing Crohn’s disease (CD). Expression of collagen, which stains blue, is higher in strictured mucosa (original magnification ×250) in comparison with non-strictured mucosa (original magnification ×250). The SMA-positive cells appear fusiform and mostly located in the pericryptal region in non-strictured mucosa (original magnification ×200), while they are less elongated and characterised by numerous projections which lengthen in the lamina propria in strictured mucosa (original magnification ×200). Data are representative of staining performed in three fibrostenosing CD patients.
Myofibroblast production of MMP-12
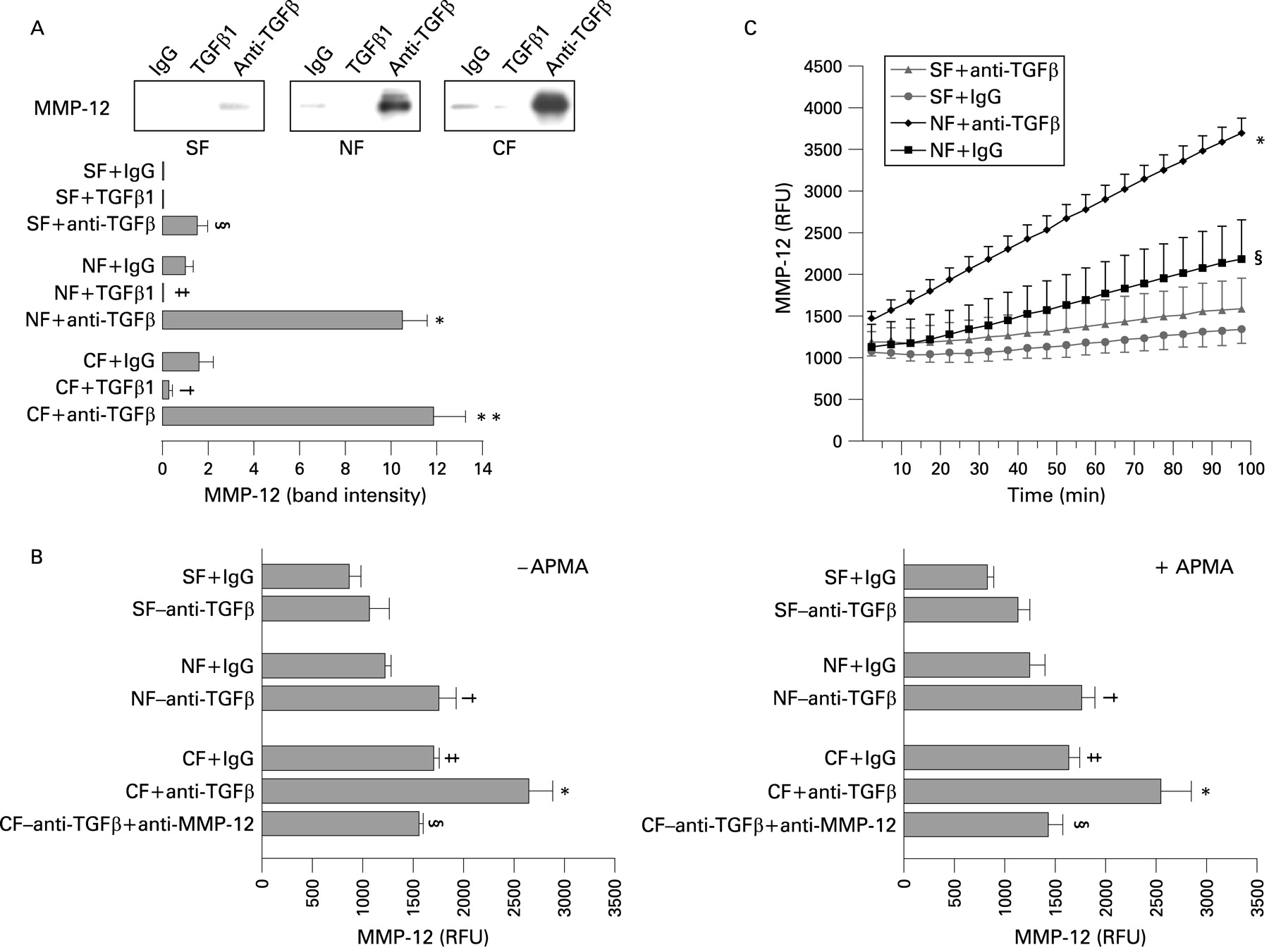
MMP-12 production was significantly increased when mucosal myofibroblasts from both fibrostenosing CD patients (n = 12) and controls (n = 11) were cultured with anti-TGFβ antibody (fig 5A). This increase was particularly evident in myofibroblasts grown from mucosa overlying non-strictured areas and control myofibroblasts. Stimulation with TGFβ1 also significantly (p<0.05) decreased MMP-12 in these latter groups of samples. However, we were not able to show any downregulatory effect of TGFβ1 on myofibroblasts from mucosa overlying strictured gut due to the low level of MMP-12 production in basal conditions. We also performed a real-time MMP-12 activity assay in the presence or absence of APMA to assess proteolytic activity in the supernatants of myofibroblasts treated with anti-TGFβ antibody (fig 5B). TGFβ blockade significantly (p<0.05) increased MMP-12 activity in the supernatants of myofibroblasts from mucosa overlying non-strictured areas, but not in myofibroblasts from mucosa overlying strictures of 12 patients with fibrostenosing CD, where the increase did not reach statistical significance. In parallel control experiments, we demonstrated that the addition of an anti-MMP-12 antibody to myofibroblasts from five control subjects significantly (p<0.05) inhibited the anti-TGFβ-induced increase of MMP-12 activity. In basal conditions, MMP-12 activity was significantly (p<0.05) higher in control mucosal myofibroblasts in comparison with myofibroblasts from mucosa overlying CD strictured gut. No significant difference was found in either the presence or absence of APMA. MMP-12 activity, measured every 5 min for 100 min, was significantly (p<0.001) increased in CD myofibroblasts from mucosa overlying non-strictured areas treated with anti-TGFβ antibody in comparison with IgG-treated cells (fig 5C). In contrast, TGFβ blockade induced only a slight increase of MMP-12 activity in the supernatants of mucosal myofibroblasts from strictured gut of five patients with fibrostenosing CD. In basal conditions, MMP-12 activity was significantly (p<0.05) higher in myofibroblasts from mucosa overyling non-strictured areas than in myofibroblasts from mucosa overlying strictured gut.

(A) Matrix metalloproteinase-12 (MMP-12) production by myofibroblasts isolated from mucosa overlying strictures (SF) and non-strictured areas (NF) from a patient with fibrostenosing Crohn’s disease (CD), and myofibroblasts from a control subject (CF), cultured with transforming growth factor β1 (TGFβ1), anti-TGFβ antibody or its isotype-matched control (rabbit immunoglobulin G (IgG)). Each example is representative of experiments performed in 12 patients with fibrostenosing CD and 11 control subjects. The lower panel shows densitometry of western blots. Results are the mean (SD) (§p<0.05 vs IgG-treated SF; *p<0.001 vs IgG-treated NF; **p<0.001 vs IgG-treated CF; ‡p<0.05 vs IgG-treated NF; †p<0.05 vs IgG-treated CF). (B) MMP-12 activity measured by real-time MMP-12 activity assay in the presence or absence of p-aminophenyl mercuric acetate (APMA) (see the Patients and methods section) and expressed in relative fluorescence units (RFU) in the supernatants of SF, NF and CF from 12 patients with fibrostenosing CD and CF from 12 control subjects, treated with anti-TGFβ antibody or IgG. In parallel control experiments, CF from five control subjects were cultured with anti-TGFβ antibody plus anti-MMP-12. No significant difference was found in either the presence or absence of APMA. Results are means (SD) (†p<0.05 vs IgG-treated NF; *p<0.05 vs IgG-treated CF; §p<0.05 vs anti-TGFβ-treated CF; ‡p<0.05 vs IgG-treated SF). (C) MMP-12 activity has been measured and recorded in RFU every 5 min for 100 min using an iQ5 real-time PCR thermal cycler (see Patients and methods section) in the supernatants of SF and NF from five patients with fibrostenosing CD treated with anti-TGFβ antibody or IgG. Results are means (SD) (*p<0.001 vs IgG-treated NF and both IgG-treated and anti-TGFβ-treated SF; §p<0.05 vs IgG-treated SF).
Myofibroblast production of MMP-3 and TIMP-1
Stimulation of myofibroblasts from mucosa overlying strictured and non-strictured gut of 12 patients with fibrostenosing CD and normal myofibroblasts from 11 control subjects with TGFβ1 or anti-TGFβ antibody or control IgG had no effect on MMP-3 expression (fig 6A). In contrast, TGFβ1 induced a significant (p<0.001) increase in myofibroblast TIMP-1 production in all the three groups of samples (fig 6B). Moreover, anti-TGFβ antibody induced no effect on TIMP-1 production in myofibroblasts from any group of patients. In basal conditions, TIMP-1 production was significantly (p<0.05) higher in CD myofibroblasts from mucosa overlying strictures than that from mucosa overlying non-strictured gut and from control mucosa.

Matrix metalloproteinase-3 (MMP-3) (A) and tissue inhibitor of metalloproteinase-1 (TIMP-1) (B) production by myofibroblasts isolated from uninflamed mucosa overlying strictures (SF) and non-strictured areas (NF) from a patient with fibrostenosing Crohn’s disease (CD), and myofibroblasts from a control subject (CF), cultured with transforming growth factor β1 (TGFβ1), anti-TGFβ antibody or its isotype-matched control (rabbit immunoglobulin G (IgG)). Each example is representative of experiments performed in 12 patients with fibrostenosing CD and 11 control subjects. Lower panels show densitometry of western blots. Results are means (SD) (*p<0.001 vs IgG-treated cells; §p<0.05 vs IgG-treated NF and CF).
Myofibroblast migration
To evaluate the possible function of different myofibroblasts involved in mucosa of CD patients, an in vitro wound-healing scratch assay was performed to assess the cell mobility/migration. Cell migration was measured as a percentage of wound repair (see Patients and methods section). We first found a significantly lower migration ability of myofibroblasts from mucosa overlying strictures in comparison with myofibroblasts from mucosa overlying non-strictured areas of 12 patients with fibrostenosing CD (p<0.05) and normal myofibroblasts from 11 control subjects (p<0.01). No significant difference was found in the migratory potential of CD myofibroblasts from non-strictured areas and control myofibroblasts (fig 7A). We then observed that myofibroblasts from mucosa overlying strictures were unresponsive to either TGFβ1 or anti-TGFβ antibody (fig 7B). In contrast, myofibroblasts from mucosa overlying non-strictured areas (fig 7C) and control myofibroblasts (fig 7D) showed significantly increased mobility in response to anti-TGFβ antibody in comparison with control IgG at 8 h (p<0.05), 16 h (p<0.001) and 24 h (p<0.001). No effect was observed when cells from mucosa overlying non-strictured areas and control cells were stimulated with TGFβ1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Migration of myofibroblasts isolated from uninflamed mucosa overlying strictures (SF) and non-strictured areas (NF) from 12 patients with fibrostenosing Crohn’s disease (CD), and myofibroblasts from 11 control subjects (CF), assessed by in vitro wound-healing scratch assay at 2, 4, 8, 16 and 24 h (see Patients and methods section). Results are means (SD) (*p<0.01 vs CF at 24 h; §p<0.05 vs CF at 24 h). (B–D) Migration of SF (B), NF (C) and CF (D) stimulated with transforming growth factor β1 (TGFβ1), anti-TGFβ antibody or its isotype-matched control (rabbit immunoglobulin G (IgG)) (§p<0.05 vs IgG-treated and anti-TGFβ-treated cells at 8 h; *p<0.001 vs IgG-treated and anti-TGFβ-treated cells at 16 and 24 h).
Myofibroblast apoptosis
To verify whether CD myofibroblasts from mucosa overlying strictures displayed an abnormal apoptotic behaviour in comparison with CD myofibroblasts from mucosa overlying non-strictured areas, apoptosis was analysed by flow cytometry using Annexin V/propidium iodide staining. As a positive control, anti-CD3/CD28-stimulated Jurkat T cells were treated with infliximab or control IgG1. No significant difference was found in the percentage of apoptosis between strictured (mean (SD) 11.0% (3.4%)) and non-strictured myofibroblasts (8.4% (3.4%)) from eight patients with fibrostenosing CD, and myofibroblasts from eight control subjects (10.4% (2.9%)). A significantly (p<0.001) higher percentage of apoptosis was found in Jurkat T cells cultured with infliximab (58.6% (8.3%)) in comparison with cells cultured with IgG1 (0.7% (0.4%)).
Myofibroblast senescence
We investigated myofibroblast senescence by detecting the senescence-associated expression of β-galactosidase activity in cell cultures at different passages (P2 and P4). The senescence-associated expression of β-galactosidase activity is present only in senescent cells and is not found in presenescent, quiescent or immortal cells.31 By performing experiments with myofibroblasts from seven patients with fibrostenosing CD and seven control subjects, we observed that cells from mucosa overlying strictures are more senescent (89.1% (4.6%) at P2 and 96.0% (3.2%) at P4; p<0.001) than cells from non-strictured mucosa (12.3% (1.7%) at P2 and 24.5% (3.1%) at P4) and normal cells (11.6% (2.2%) at P2 and 23.0% (4.3%) at P4). No significant difference was found between cells from non-strictured areas and control cells. As shown in table 2, high passage control and non-strictured myofibroblasts, having the same senescence as strictured myofibroblasts, show TGFβ transcript levels, collagen production, MMP production and Smad signalling comparable with those of low passage control and non-strictured myofibroblasts.
DISCUSSION
Intestinal strictures in CD are the end result of chronic transmural inflammation and dysregulated wound repair which cause excessive deposition of ECM. Abnormal contraction of ECM proteins leads to scar formation and ultimately intestinal obstruction.3 While the mechanisms of inflammation in CD have been extensively investigated, knowledge of stricture pathogenesis is limited because of the absence of a good mouse model and the difficulty in obtaining non-surgical samples.1 2 32 In this study we have taken a different approach and have used conventional or CT enteroclysis to identify areas of strictures in the bowel and then sample macroscopically normal mucosa above these areas. Likewise we were also able to identify areas without obvious strictures and sample mucosa above these areas. Importantly, macroscopically and microscopically the samples we took showed either no inflammation or minimal inflammation, so any confounding effects of inflammation on MMPs and TGFβ signalling were minimised.
Our studies identified several major differences between mucosa above strictures and that of mucosa above no apparent strictures. First, there was an increase in TGFβ transcripts in the former, and this was associated with the molecular signature (pSmad2–3) of increased TGFβ signalling, low Smad7. Secondly, in the former there was less MMP-3 and MMP-12 protein, and more TIMP-1. Thirdly, in myofibroblasts isolated from mucosa overlying strictured gut there was more collagen production and more TGFβ transcripts, and the cells appeared more responsive to activation of Smad2–3 by exogenous TGFβ1. Fourthly, while TGFβ itself inhibited MMP-12 production by myofibroblasts from all groups of patients, neutralising anti-TGFβ antibody led to a dramatic increase in MMP-12 production and activity by myofibroblasts from mucosa overlying non-strictured CD gut or normal mucosa, but barely any effect with myofibroblasts from mucosa overlying strictures. This effect was not seen for MMP-3 or TIMP-1 though TGFβ was a strong inducer of TIMP-1 in all myofibroblasts.
To try to put all these observations together, it would appear that essentially normal mucosa overlying strictured gut is different from that overlying non-strictured gut in terms of molecules associated with fibrosis, namely increased TGFβ and TGFβ signalling, decreased MMP production and increased TIMP-1. These effects are recapitulated to some extent in isolated myofibroblasts, though differences were seen in the regulation of MMP-3 and MMP-12 by TGFβ.
The first observation worthy of discussion is the elevated TGFβ transcripts, Smad signalling and collagen expression in normal mucosa overlying strictured bowel. At least part of this increase is probably from myofibroblasts, but the contribution of other cell types cannot be discounted. One possibility is that this represents the “shadow” of the fibrotic process ongoing in the deeper layers. Vallance et al33 have demonstrated that epithelial expression of active TGFβ in mouse colon leads to fibrosis in the deeper layers, presumably because it initiates a cascade which is communicated intercellularly through the mucosa. It is therefore possible that in this situation, the reverse is happening. Alternatively, given the dynamic nature of the CD mucosal lesions, the samples we took, while histologically normal, may have been inflamed in the past and the changes are again the molecular shadow of a healing mucosa. A final and highly speculative possibility is that the changes we have seen in the mucosa are primary and that the evolution into fibrosis in the deeper layers is because this tissue does not have the potent anti-inflammatory and antifibrotic capacity of the mucosa.
The second point worthy of discussion is the low level of matrix-degrading enzymes and high TIMP-1 seen in mucosa overlying strictured gut. This is somewhat reminiscent of the situations in collagenous colitis5 and diverticular disease6 where collagen synthesis and TIMP-1 are elevated, though MMP production is normal. However, it again points to the mucosa overlying fibrotic CD as having a reduced capacity to degrade ECM, one of the key features of matrix accumulation, fibrosis and scarring. MMPs are neutral endopeptidases involved in regulating the process of tissue injury and wound healing in the gut through the modulation of ECM degradation and deposition.12 34 The major role of MMPs in mediating mucosal damage in chronic intestinal inflammation has been widely highlighted by us and others in recent years.16 21–24 35 MMPs act to degrade many ECM proteins involved in tissue fibrosis, such as collagen, laminin and fibronectin.12 Hence, it is conceivable that reduced activity may facilitate the development of fibrotic lesions. The two MMPs we looked at, MMP-3 which degrades mostly type IV collagen and proteoglycans,12 and MMP-12, which has potent elastase activity,36 were both markedly decreased in the mucosa overlying strictured gut. However, when we isolated myofibroblasts from the tissues, while the low MMP-12 production was maintained, MMP-3 production really showed no difference between any of the groups, suggesting either selection for myofibroblast subsets when the cell lines were established or changes due to the in vitro situation. Among MMPs, the functional role of MMP-12 in CD has been rather neglected, since the elastin, a major substrate for MMP-12,36 is not abundantly expressed in the gut.37 However, MMP-12 is also capable of degrading a broad spectrum of other ECM components, including type IV collagen, fibronectin, laminin and vitronectin,38 39 all of which are present in CD strictures.1 3 40
We also carried out somewhat complex experiments to look at the effects of exogenous TGFβ on Smad signalling, and MMP and TIMP production by myofibroblasts and the effects of neutralising TGFβ, to determine if autocrine production of TGFβ by myofibroblasts played any role in their function. As expected, neutralising TGFβ abolished Smad signalling in all myofibroblasts,41 but, interestingly, addition of exogenous activated TGFβ induced a greater response in myofibroblasts from mucosa overlying strictured gut. One might wonder why this is the case since the same cells appear to be making lots of TGFβ transcripts; however, the critical thing is the amount of active TGFβ that is present and not the total amount, so these might be unrelated events. Overall, however, it does suggest that myofibroblasts from mucosa overlying CD strictures are hyper-responsive to TGFβ.
Interesting results were also obtained with MMP-12. Equivalent to the situation in vivo, myofibroblasts from mucosa overlying strictures made little MMP-12 and consequentially addition of exogenous active TGFβ had no effect, though it is well established that TGFβ is a potent inhibitor of MMP-12.16 42 However, when TGFβ was neutralised, while there was only a modest increase in MMP-12 production by myofibroblasts from mucosa overlying strictures in control myofibroblasts and CD myofibroblasts from mucosa overlying non-strictured areas, there was large increase in MMP-12, confirmed by both immunoblotting and direct proteolytic activity. This suggests that there may be epigenetic silencing of the MMP-12 locus in myofibroblasts from mucosa overlying strictures to explain the persistence of the effect in vitro. However, when we looked at MMP-3, we essentially found no differences between all groups of myofibroblasts. We cannot explain the difference between the in vivo and in vitro data unless the MMP-3 is being made by non-myofibroblasts, or the cells changed function when they were passaged.
Development of fibrosis has been associated with changes in the migratory potential of mesenchymal cells in a number of disorders.43 44 Myofibroblast migration is an important mechanism of intestinal wound healing.45 In chronic inflammation, persistent mucosal ulceration is accompanied by insufficient wound repair due to a reduced migration of intestinal myofibroblasts.16 46 We here observed a defect in the migratory potential of myofibroblasts derived from mucosa overlying CD strictures. Increased deposition of ECM proteins due to the local increase in the number of CD myofibroblasts with reduced dispersing potential may provide a pathophysiological explanation for this reduced migration, and indeed in our experiments the cells from mucosa overlying strictures made more collagen. These results are also somewhat similar to the results of Leeb et al46 who showed that inflammatory bowel disease myofibroblasts migrated less than control myofibroblasts. Preliminary results by Leeb et al46 also showed that TGFβ1 enhanced myofibroblast migration, so we determined whether blocking TGFβ might lower myofibroblast migration. We found, however, that TGFβ neutralization significantly enhanced the migration of non-strictured myofibroblasts. Interestingly, we observed that cells from mucosa overlying strictures were more senescent than cells isolated from non-strictured mucosa. This is in keeping with the fact that strictured myofibroblasts grow more slowly than non-strictured or normal myofibroblasts. Additionally, we found that the behaviour of control and non-strictured myofibroblasts is not related to the senescence state as high passage control and non-strictured myofibroblasts, which have the same senescence as strictured myofibroblasts, show a TGFβ/collagen/MMP/Smad profile comparable with that of low passage cells. No difference was found in the percentage of apoptosis between strictured and either non-strictured or control myofibroblasts.
In conclusion, the abundance of TGFβ and TIMP-1, and the lack of MMP-3 and MMP-12, in mucosa overlying CD strictures, together with the role of TGFβ blockade in reversing MMP-12 and upregulating TIMP-1 myofibroblast production, suggest that aspects of the fibrogenic process may be demonstrated in essentially normal mucosa overlying strictured gut in CD. There are two obvious implications of this work, first that the mucosal changes are a shadow of events deeper in the bowel, or, alternatively, that it is the changes in the mucosa which are driving the changes in the deeper bowel. It seems quite clear that overt fibrosis is a very rare event in the mucosa, so the development in the deeper layers of the gut may in part be due to the inability of that tissue to inhibit fibrosis compared with the mucosa.
Acknowledgments
The authors would like to thank Dr Peter Lackie and Dr David Johnston for their advice in setting up the wound scratch assay, Claire Webb for her excellent technical support, and all patients who agreed to give biopsies and tissue samples, without whom this study would not have been possible. This project was supported in part by the European Union framework programme 7 collaborative project grant IPODD (call: FP7-Health-2007-A), the Broad Medical Research Programme, USA and the Innovation Fund, Hope, Southampton, UK.
REFERENCES
Footnotes
Competing interests: None.
Ethics approval: Appropriate local Ethics Committee approval was obtained for this study
Patient consent: Informed consent was obtained in all cases.
Linked Articles
- Digest