Article Text

Abstract
Nucleotide oligomerisation domain 2 (NOD2) mutations are associated with susceptibility to Crohn's disease and graft-versus-host disease, two human disorders related with dysfunctions of Peyer's patches (PPs). In Nod2−/− mice transcellular permeability and bacterial translocation are increased in PPs. In this study, we show that both anti-CD4+ and anti-interferon γ (anti-IFNγ) monoclonal antibodies abrogate this phenotype and reduce the expression of tumour necrosis factor (TNF) receptor 2 and the long isoform of myosin light chain kinase, thus demonstrating that immune T cells influence the epithelial functions. In turn, intraperitoneal injection of ML-7 (a myosin light chain kinase inhibitor) normalises the values of CD4+ T cells, IFNγ and TNFα. This reciprocal cross-talk is under the control of the gut microflora as shown by the normalisation of all parameters after antibiotic treatment. Toll-like receptor 2 (TLR2) and TLR4 expression were increased in Nod2−/− mice under basal conditions and TLR2 and TLR4 agonists induced an increased transcellular permeability in Nod2+/+ mice. Muramyldipeptide (a Nod2 agonist) or ML-7 was able to reverse this phenomenon. It thus appears that Nod2 modulates the cross-talk between CD4+ T cells and the epithelium recovering PP and that it downregulates the pro-inflammatory effect driven by the ileal microflora, likely by inhibiting the TLR pathways.
- Nod2
- Peyer's patch
- MLCK
- epithelial permeability
- gut microflora
- Crohn's disease
- graft-versus-host disease
- IBD
- IBD basic research
- immune response
- CARD15
- caspase recruitment domain 15
- CD
- Crohn's disease
- DC
- dendritic cell
- FAE
- follicle-associated epithelium
- GALT
- gut-associated lymphoid tissue
- GVHD
- graft-versus-host disease
- LFs
- lymphoid follicles
- LPS
- lipopolysaccharide
- MDP
- muramyl dipeptide
- MLCK
- myosin light chain kinase
- NLRC2
- natch Leucine-rich-repeat Card 2
- NOD2
- nucleotide oligomerisation domain 2
- PGN
- peptigoglycan
- PP
- Peyer's patches
- Th1/2
- T helper type 1/2
- TLR
- toll-like receptor
- TNFR
- TNF receptor
Statistics from Altmetric.com
- Nod2
- Peyer's patch
- MLCK
- epithelial permeability
- gut microflora
- Crohn's disease
- graft-versus-host disease
- IBD
- IBD basic research
- immune response
- CARD15
- caspase recruitment domain 15
- CD
- Crohn's disease
- DC
- dendritic cell
- FAE
- follicle-associated epithelium
- GALT
- gut-associated lymphoid tissue
- GVHD
- graft-versus-host disease
- LFs
- lymphoid follicles
- LPS
- lipopolysaccharide
- MDP
- muramyl dipeptide
- MLCK
- myosin light chain kinase
- NLRC2
- natch Leucine-rich-repeat Card 2
- NOD2
- nucleotide oligomerisation domain 2
- PGN
- peptigoglycan
- PP
- Peyer's patches
- Th1/2
- T helper type 1/2
- TLR
- toll-like receptor
- TNFR
- TNF receptor
Introduction
Caspase recruitment domain 15 (CARD15) also known as nucleotide oligomerisation domain 2 (NOD2) and NOD-LRR-CARD-2 (NLRC2) belongs to a family of genes involved in innate immunity.1 Like toll-like receptors (TLR) Nod2 is considered as a sensor of bacterial components. It can be activated by muropeptides, which are components of the bacterial cell wall. Nod2 signalling can interact with TLR signalling. Depending on the experimental conditions Nod2 may exert synergistic or inhibitory effects on TLR pathways.2–5
NOD2 polymorphisms have been associated with Crohn's disease,6 7 a chronic relapsing inflammatory bowel disease (IBD) with mucosal ulcerations of the digestive tract.8 In Crohn's disease, 30–50% of patients exhibit one or more Nod2 genetic variations, which are usually considered as loss of function mutations. As a consequence, Crohn's disease is often seen as an immune deficiency towards bacteria present in the gut. However, the exact mechanisms by which Nod2 mutations are able to induce Crohn's disease lesions are still subject to debate.
NOD2 has also been associated with mortality and graft-versus-host disease (GVHD) after bone marrow transplantation.9 10 GVHD is associated with increased intestinal permeability and experimental models indicate that the primacy of gastrointestinal damage results in bacterial translocation followed by T cell activation and an increased cytokine release.11 12 The incidence of severe GVHD rises from 18% in donor/recipient pairs without any NOD2 variants to 37% in pairs with either donor or recipient mutations and 22–55% in pairs with both members mutated.10 13 NOD2 mutations seem to play a symmetric role in both donors and recipients, suggesting that NOD2 impairs function of both non-immune cells of the host and circulating immune cells of the graft.
Crohn's disease and GVHD lesions require gut-associated lymphoid formations known as lymphoid follicles (LFs).14 15 LFs are encountered in the colon where they are isolated and in small bowel where they are grouped forming Peyer's patches (PPs). We have recently shown that Nod2 invalidation in mice induces an hypertrophy and a hyperplasia of PPs after birth.16 Crohn's disease and GVHD are characterised by a T helper 1 (Th-1) immune response with elevated rates of interferon γ (IFNγ) and tumour necrosis factor α (TNFα) in the mucosa.17 In both disorders, the intestinal barrier is disrupted as shown by an excessive gut permeability of macromolecules and bacteria.18 19 However, the link between Nod2 deficiency and the mucosal abnormalities observed in Crohn's disease or GVHD has not been fully explored. The aim of the present study was to dissect this link in a mouse model invalidated for NOD2.
Material and methods
Animals and experimental protocols
C57BL/6 Nod2+/+, Nod2−/− and Nod2mut/mut mutated mice were generated in the animal facility at Robert Debré Hospital.16 20 All mice were housed in pathogen-free conditions with free access to food and water. Pathogen-free conditions were monitored every 6 months in accordance with the full set of FELASA high standards recommendations.
In vivo depletion of CD4+ T cells was obtained by intraperitoneal injections of 100 μg purified GK1.5 (anti-L3T4 (CD4+) monoclonal antibody (Pharmingen, Heidelberg, Germany) at both 96 and 24 h before experimentation.21 For in vivo depletion of IFNγ, mice were intraperitoneally injected with 200 μg of a rat anti-mouse anti-IFNγ Ab XMG1.2 (Pharmingen) 24 h before experimentations.22 Myosin light chain kinase (MLCK) inhibition was obtained by intraperotoneal injection twice daily of ML-7 (2 mg/kg; Sigma, Saint Quentin, France), a MLCK inhibitor, for 4 days before experiments.23 Antibiotic protocols consisted in the administration of 1.0 g/l ampicillin and 0.5 g/l neomycin (Sigma) in drinking water for 12 days.23
Peyer's patches and spleen studies
Small intestine from Nod2−/− and Nod2+/+ mice were removed and fixed in 4% phosphate-buffered formalin overnight. Samples were then stained with 0.5% methylene blue and decolourised in fresh 2% acid acetic. Then PP numbers were counted in a blind fashion.
Cell suspensions from PPs were prepared by pressing ileal PP with a syringe piston.16 Cells from spleens were isolated using the same procedure with an additional step of erythrocyte lysis (Gey's solution). After centrifugation, cells were re-suspended in Dulbecco's modified Eagle's medium (DMEM) and submitted to flow cytometry analyses on a FACScalibur (Becton-Dickinson, Le Pont-de-Claix, France), and analysed by Cell Quest 3.3 (Becton Dickinson). Monoclonal antibodies used to stain cell suspensions were purchased from BD (Pharmingen): PE-Cy7-anti-CD3 (145-2C11), PE-Cy5-anti-CD4 (H129.19), fluorescein isothiocyanate (FITC)–anti-CD8 (53-6.7).
PPs and spleens from Nod2+/+ and Nod2−/− mice were removed, and the concentration of proteins was determined using a commercial kit (Biorad, Marnes la Coquette, France). TNFα and IFNγ levels were determined by ELISA assays (BD Biosciences, Pharmingen, Hedelberg, Germany) according to the manufacturer's instructions.
Ussing chamber experiments
Biopsies from PPs were placed in a chamber exposing 0.196 cm2 of tissue surface to 1.5 ml of circulating oxygenated Ringer solution at 37°C. PP transcellular permeability were assessed by measuring steady-state (from 1 to 2 h) mucosal-to-serosal flux of 40 kDa FITC–dextran (Sigma). Bacterial translocation was studied using chemically killed fluorescein-conjugated Escherichia coli or Staphylococcus aureus BioParticles (Molecular Probes, Leiden, The Netherlands) at a final concentration of 1.107 cfu/ml in the mucosal reservoir.
To investigate the role of TLR2/4 in the increase PP transcellular permeability, ultrapure and contaminant-free Pam3CSK4 (TLR2 agonist; Invivogen, Steinheim, Germany), PGN (TLR2 agonist; Fluka, San Diego, California, US) or lipopolysaccharide (LPS (TLR4 agonist; Sigma) were added into the luminal side of the Ussing chamber at final concentration 20 μg/ml. To investigate the effect of Nod2 stimulation, mice were pre-treated intraperitoneally with MDP (100 μg/mouse/day; Sigma) for two consecutive days before experimentation and Ussing chamber experiments were performed after adding 10 μg/ml of MDP into the luminal side. For MLCK inhibition, mice were pre-treated by ML7 (see above) and 20 μg/ml of ML-7 were added in the Ussing chamber.
Measurement of in vivo transcellular permeability
Transcellular permeability was assessed by oral administration of fluorescein isothiocyanate-labelled dextran 40 kDa (FITC–dextran; Sigma). Mice were gavaged with FITC–dextran (12 mg/300 μl/mouse) 5 h prior to sacrifice. Whole blood FITC–dextran concentration was determined by a fluorimeter and transcellular permeability was expressed as the mean whole blood FITC–dextran concentration in ng/ml.
Immunohistochemistry of dendritic cells and apoptotic cells
PPs were removed and washed in cold phosphate-buffered saline (PBS), then samples were embedded in Tissue Tek medium (Euromedex, Souffelweyersheim, France). Cryostat sections (5 μm) were post-fixed with acetone (10 min, −20°C) and washed in PBS to eliminate Tissue Tek. Endogen peroxidases were blocked with 3% H2O2 (DAKO, Carpinteria, California, USA) and slides were incubated for 60 min with primary antibodies (purified hamster anti-mouse CD11C, dilution: 1/100; BD Pharmingen). A secondary antibody (anti-hamster) was then applied for 30 min, followed ABC kit (Vector Laboratories, Burlingame, California, USA) for 30 min. For detection, liquid 3,3′-diaminobenzidine (DAB) and Substrat (DAKO) was added for 10 min. Counts of positive CD11c cell in PPs were performed in a blind fashion.
After 2 h in the Ussing chamber, PPs were removed and washed in cold PBS. Then, PP samples were fixed in 4% phosphate-buffered formalin and embedded in paraffin blocks and cut into 5 μm sections. Briefly, 5 μm deparaffinised sections were subjected to a heat-induced antigen recovery in sodium citrate buffer solution pH 6. Endogen peroxidases were blocked with 3% H2O2 (DAKO) and slides were incubated for 30 min with primary antibodies (rabbit polyclonal antibody against cleaved caspase-3 (Asp 175, dilution: 1/100; Cell Signaling Technology, Ozyme, Beverly, Massachusetts, USA). A biotin-labelled secondary antibody was then applied for 30 min, followed by avidin–biotin–peroxidase conjugate for 30 min (Vector Laboratories). For detection, peroxidase enzyme substrate 3,3′-diaminobenzidine was added to yield a brown reaction product. The slides were counterstained with Harris haematoxylin. Counts of positive caspase-3-cleaved cells in PPs were performed in a blind fashion.
Ileal bacterial counts
Ileum was removed and ileal content was collected using sterile water (Biorad). Then, ileal content was homogenised and serial dilutions of each aliquot (50 μl) were plated onto five selective gelose plates (URI 4, Drigalski, Columbia ANC + 5% of sheep blood, Chapman and Coccosel). Plates were incubated for 24 h at 37°C under aerobic condition and the number of colony forming units was counted and expressed as cfu/mg of ileal content.
Real time reverse transcription-polymerase chain reaction
After extraction by the NucleoSpin RNA II Kit (Macherey-Nagel, Hoerdt, France), total RNAs were converted to cDNA using random hexonucleotides and then used for RT-PCR. We conducted PCR with QuantiTect SYBR Green PCR Kit (Applied, Courtaboeuf, France) using sense and antisense primers specific for G3PDH, the long MLCK isoform (specifically expressed by epithelial cells), tumour necrosis factor receptor 1 (TNFR1), TNFR2, TLR2 and TLR4 (sequence are shown in supplementary table 1). After amplification, we determined the threshold cycle (Ct) to obtain expression values of 2−ΔCt.
Statistical analyses
Values are expressed as mean ± SEM. Statistical analysis were performed using GraphPad Prism 4.00 software package for PC. Comparisons were performed by the unpaired Student t test. A value of p < 0.05 was considered as statistically significant.
Results
CD4+ T cells and IFNγ trigger PP barrier dysfunction in Nod2−/− mice
As previously reported,16 CD4+ T–cells were increased in the PPs of Nod2−/− mice (figure 1A). In order to define their role, Nod2+/+ and Nod2−/− mice were treated with anti-CD4+ antibodies. CD4+ T cell depletion was effective in both PPs and spleen as shown in figure 1A and supplementary figure 1A. It was associated with a decrease of IFNγ and TNFα tissue concentration (figure 1B,C and supplementary figure S1B,C). Similarly, CD4+ T cells abrogated the increase of PP transcellular permeability as well as S aureus translocation in Nod2−/− mice (figure 1D,E). In addition, after CD4+ T cell depletion these parameters were similar in Nod2+/+ and Nod2−/− mice (figure 1D,E). We thus concluded that CD4+ T cells play a major role in the whole phenotype observed in case of Nod2 deficiency.

CD4+ T cell depletion restores a normal phenotype of Peyers's patches (PPs) from Nod2−/− mice. Flow cytometry, ELISA and Ussing-chamber experiments were performed on PPs from Nod2−/− and Nod2+/+ mice. (A) T cell composition of PPs. Each plot shows staining after gating on live CD3+ T cells. (B and C) PP levels of interferon γ (IFNγ) and tumour necrosis factor α (TNFα). (D) Transcellular permeability was analysed by fluorescein isothiocyanate (FITC)–dextran flux. (E) Bacterial translocation of Staphylococcus aureus. Data represent the mean ± SEM of eight mice per group.*p <0.05, **p <0.01 and ***p <0.001, significantly different from Nod2+/+ under basal conditions; ††p <0.01 and †††p <0.001, significantly different from Nod2−/− under basal conditions. KO, knock-out; WT, wild-type.
CD4+ T cells are known to produce IFNγ which is a key factor in the Th1 response and we have previously shown that IFNγ levels are increased in Nod2−/− mice.16 In order to test the role of IFNγ on the phenotype, we then treated mice with anti-IFNγ antibodies. In PPs and spleen, IFNγ concentrations measured by ELISA methods, were dramatically reduced in both Nod2−/− and Nod2+/+ mice (figure 2A,B). In parallel, the transcellular permeability and the bacterial ingress were also decreased in PP from Nod2−/− mice (figure 2C,D). A similar but less dramatic effect was observed in Nod2+/+ mice (figure 2C,D). As a result, after IFNγ depletion, epithelial permeability and bacterial translocation rates were similar in Nod2−/− and Nod2+/+ mice. Thereby IFNγ modulate the intestinal function in Nod2−/− mice.

Interferon γ (IFNγ) contributes to disruption of the function of Peyer's patches (PPs) in Nod2−/− mice. ELISA and Ussing-chamber experiments were performed on PPs and spleens from Nod2−/− and Nod2+/+ mice. (A and B) IFNγ levels in PPs and spleens. (C) Transcellular permeability was analysed by fluorescein isothiocyanate (FITC)–dextran flux. (D) Bacterial translocation of Staphylococcus aureus. Data represent the mean±SEM of eight mice per group. *p <0.05, **p <0.01 and ***p <0.001, significantly different from Nod2+/+; ††p <0.01 and †††p <0.001, significantly different from Nod2−/− under basal conditions. KO, knock-out; WT, wild-type.
In other animal models, IFNγ and T cells have been reported to disrupt epithelial barrier integrity by upregulating TNFR2 and MLCK expression.23 24 We thus investigated the mRNA expression of TNFR1, TNFR2 and MLCK. Under basal condition and after CD4+ or IFNγ depletion, no significant differences were shown for TNFR1 mRNA expression in PPs (figure 3A). In contrast, an increased expression of TNFR2 and the long isoform of MLCK were seen in Nod2−/− mice compared to controls (figure 3B,C). After CD4+ or IFNγ depletion, TNFR2 mRNA expression decreased in PP from Nod2+/+ and Nod2−/− mice with a normalisation of TNFR2 mRNA expression in Nod2−/− mice. IFNγ or CD4+ depletions were able to reverse this phenotype while they did not affect MLCK expression in Nod2+/+ mice. We thus concluded that CD4+ and IFNγ modulate the MLCK expression in epithelial cells recovering PPs.
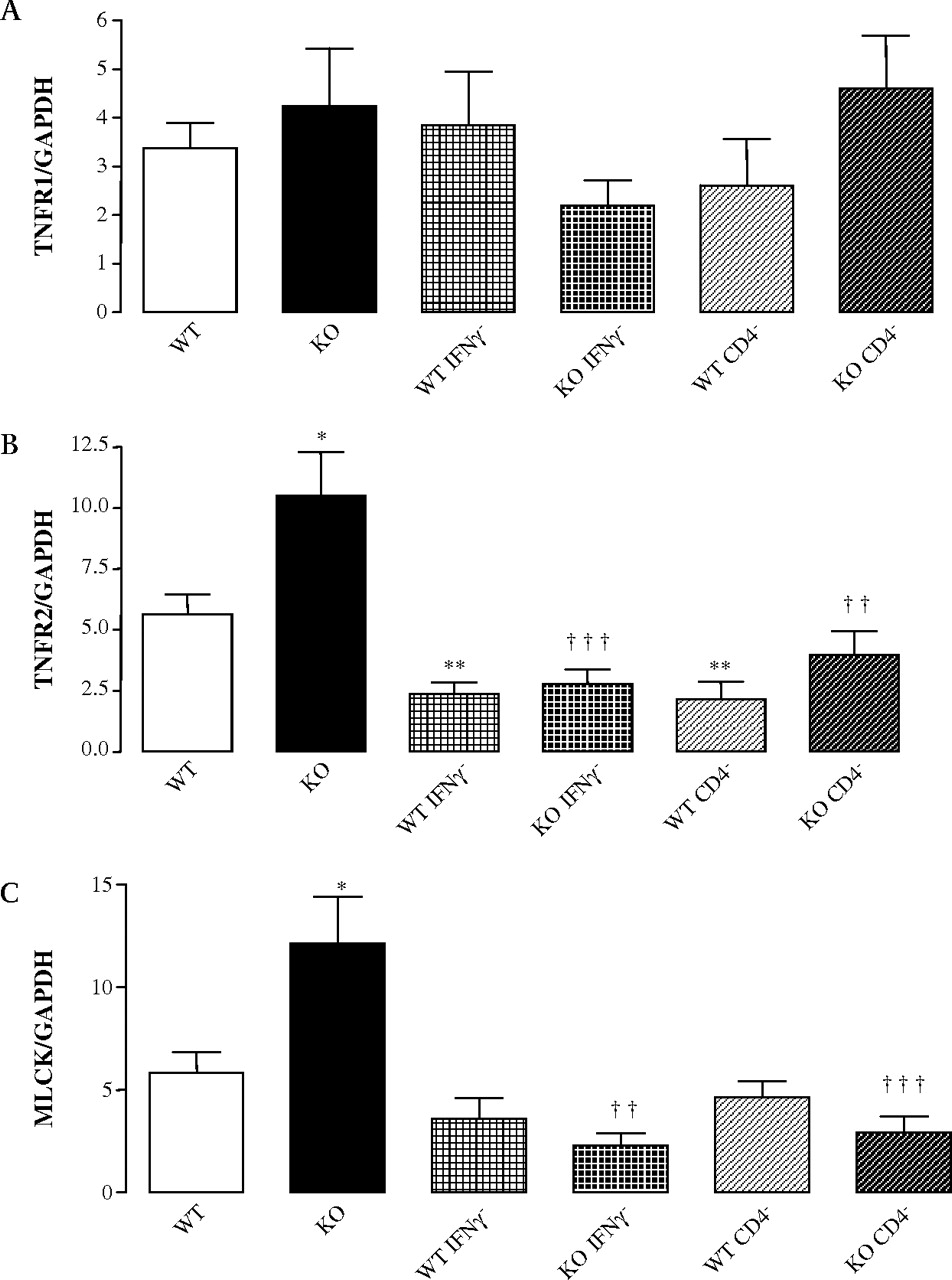
CD4+ T cells and IFNγ depletion abrogate the increased mRNA expression of TNFR2 and MLCK in PPs from Nod2−/− mice. Real-time PCR experiments were performed on PPs from Nod2−/− and Nod2+/+ mice. (A–C) TNFR1, TNFR2 and MLCK mRNA expression. Data represent the mean±SEM of eight mice per group. *p <0.05 and **p <0.01 significantly different from Nod2+/+ under basal conditions; ††p <0.01 and †††p <0.001, significantly different from Nod2−/− under basal conditions. IFNγ, interferon γ; KO, knock-out; MLCK, myosin light chain kinase; PCR, polymerase chain reaction; PPs, Peyer's patches; TNFR, tumour necrosis factor receptor. WT, wild-type.
Epithelial MLCK is involved in PP homeostasis in Nod2−/− mice
As MLCK has been reported to disrupt the epithelial integrity23–25 we further investigated its role by treating the animals with ML-7, a MLCK inhibitor. In Nod2+/+ mice, ML-7 slightly reduced the ingress of S aureus through the epithelium recovering PP while it did not affect the transcellular permeability as well as the bacterial passage of E coli (figure 4A–C). In contrast, in Nod2−/− mice, the PP permeability and the bacterial ingresses (E coli and S aureus) were dramatically reduced by ML-7 treatment with a normalisation of these parameters compared to control mice. We concluded that MLCK plays a key role in the observed epithelial dysfunction in Nod2−/− mice.

MLCK participates in the disruption of PP function in Nod2−/− mice. Ussing-chamber experiments were performed on PPs from Nod2−/− and Nod2+/+ mice. (A) Transcellular permeability was analysed by FITC–dextran flux. (B and C) Bacterial translocation of Staphylococcus aureus or Escherichia coli. Data represent the mean±SEM of eight mice per group. **p <0.01 and ***p <0.001, significantly different from Nod2+/+ under basal conditions; †p <0.05, ††p <0.01 and †††p <0.001, significantly different from Nod2−/− under basal conditions. FITC, fluorescein isothiocyanate; KO, knock-out; MLCK, myosin light chain kinase; PPs, Peyer's patches. WT, wild-type.
We further explored the impact of the observed epithelial abnormalities on the mucosal inflammation. ML-7 treatment decreased the percentage of CD4+ T cells in Nod2−/− mice while it did not affect the cellular composition in Nod2+/+ mice (figure 5A). Indeed, after ML-7 treatment, PP from Nod2+/+ and Nod2−/− mice exhibited similar rates of CD4+ T cells. In comparison, ML-7 treatment did not change the T cell composition in spleen (supplementary figure S2). Comparable results were obtained in terms of cytokine levels in the PPs. ML-7 treatment did not modify the IFNγ and TNFα levels in Nod2+/+ mice (figure 5B,C). In contrast, it decreased IFNγ and TNFα tissue concentrations in Nod2−/− mice, turning these values to normal ranges. Here too, ML-7 did not affect the studied parameters in the spleen (supplementary figure S2). Finally, ML-7 decreased and restored a normal expression of TNFR2 and MLCK but did not change TNFR1 mRNA expression in PP from Nod2−/− mice (figure 5D–F). Consequently, we concluded that MLCK plays a major role in the PP phenotype of Nod2−/− mice.

MLCK is involved in the regulation of PP homeostasis in Nod2−/− mice. Flow cytometry, ELISA and real-time PCR experiments were performed on PPs from Nod2−/− and Nod2+/+ mice. (A) T cell composition was investigated using antibodies to CD3, CD4, CD8. Each plot shows staining after gating on live CD3+ T cell. (B and C) Levels of IFNγ and TNFα. (D–F) TNFR1, TNFR2 and MLCK mRNA expression. Data represent the mean±SEM of eight mice per group. *p <0.05 and **p <0.01, significantly different from Nod2+/+ under basal conditions; †p <0.05, ††p <0.01 and †††p <0.001 significantly different from Nod2−/− under basal conditions. IFNγ, interferon γ; KO, knock-out; MLCK, myosin light chain kinase; PPs, Peyer's patches; TNFα, tumour necrosis factor α; TNFR, TNF receptor; WT, wild-type.
Antibiotic treatment reverses the phenotype of Nod2−/− mice
Crohn's disease and GVHD lesions are related to an abnormal response of the host towards bacteria. We thus analysed the role of the gut microflora in the observed phenotype. Both Nod2+/+ and Nod2−/− mice were treated by antibiotics (ABTs). ABT treatment almost totally suppressed the aerobic microflora and only a small population of Staphylococcus spp. persisted at a comparable level in Nod2+/+ and Nod2−/− mice (supplementary table 2). While treatment with ABTs did not change the number of PP in Nod2+/+ mice, it significantly decreased the number of PP in Nod2−/− mice (figure 6A). In addition, ABTs reduced the number of immune cells per PP in Nod2−/− and Nod2+/+ mice (figure 6B). In contrast, the number of splenocytes was not affected by ABT treatment in either mice strain (supplementary figure S3).

Ileal microflora disrupt PP homeostasis in Nod2−/− mice. (A) PP count on the whole intestines of Nod2−/− and Nod2+/+ mice. (B) Number of immune cells per PP in basal conditions and after ABTs. (C) T cell composition of PPs was investigated using antibodies to CD3, CD4 and CD8. Each plot shows staining after gating on live CD3+ T cells. (D and E) Levels of IFNγ and TNFα. Data represent the mean±SEM of eight mice per group. *p <0.05, **p <0.01 and ***p <0.001, significantly different from WT under basal conditions; †p <0.05, ††p <0.01 and †††p <0.001, significantly different from Nod2−/− under basal conditions and; #p <0.05 significantly different Nod2+/+ after ABTs. ABTs, antibiotics; IFNγ, interferon γ; KO, knock-out; PPs, Peyer's patches; TNFα, tumour necrosis factor α; WT, wild-type.
ABT strongly reduced the percentage of CD4+ T cells in PPs of both Nod2+/+ and Nod2−/− mice (figure 6C) demonstrating that the ileal microflora modulates the percentage of CD4+ T cells in PPs. In contrast, ABTs did not alter the percentage of CD4+ T cells in spleens from Nod2+/+ and Nod2−/− mice (supplementary figure S3). ABTs also decreased the levels of IFNγ and TNFα in PPs of Nod2−/− mice (figure 6D,E). In parallel, in PPs from Nod2−/− ABTs decreased TNFR2 and MLCK mRNA expression (figure 7A–C). They reduced the permeability and bacterial passage through PPs (figure 7D–F).

Ileal microflora alters TNFR2 and MLCK expression and changes PP function in Nod2−/− mice. Real-time PCR and Ussing-chamber experiments were performed on PPs from Nod2−/− and Nod2+/+. (A–C) TNFR1, TNFR2 and MLCK mRNA expression. (D) Transcellular permeability were analysed by FITC−dextran flux in PPs. (E and F) Translocation of Staphylococcus aureus and Escherichia coli. Data represent the mean±SEM of eight mice per group. *p <0.05, **p <0.01 and ***p <0.001, significantly different from Nod2+/+ under basal conditions; †p <0.05, ††p <0.01 and †††p <0.001, significantly different from Nod2−/− under basal conditions. ABT, antibiotic; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; KO, knock-out; MLCK, myosin light chain kinase; PPs, Peyer's patches; TNFR, tumour necrosis factor receptor; WT, wild-type.
Together these results show that suppression of the intestinal microflora is able to fully reverse the phenotype of Nod2−/− mice. In contrast, ABT treatment had only limited effects on the PP phenotype of Nod2+/+ mice and no changes were observed in the spleen of both mouse strains after ABT (supplementary figure S3). Finally, comparable results were obtained when using ABT protocols containing metronidazole (antibiotic spectrum against anaerobic bacteria) in addition to ampicillin and neomycin (data not shown).
Nod2 downregulates the increased PP permeability driven by TLR2/4 agonists
Nod2 plays a crucial role in the immune tolerance to bacteria by decreasing the production of inflammatory cytokines in response to TLR2/4.2 3 5 Consequently, we investigated whether this negative cross-talk between TLR and NOD2 may contribute to the increased permeability observed in PP from Nod2−/− mice.
Under basal condition, PP from Nod2−/− mice exhibited an increased mRNA expression of TLR2 and TLR4 in comparison with controls (figure 8A,B). CD4+ depletion, IFNγ depletion, ABT treatment or ML-7 treatment were all able to decrease TLR2 and TLR4 expressions in PP from Nod2−/− mice, while they had only a limited effect in Nod2+/+ mice. This observation suggests that TLR2 and TLR4 over-expression in Nod2−/− mice is secondary to the abnormal PP phenotype. Next, we studied the effect of agonists for TLR2 (Pam3CSK4 and PGN) and/or TLR4 (purified LPS) on PP permeability. These molecules do not alter the number of apoptotic cells of PPs mounted in Ussing chambers (supplementary figure S5). Any of the studied TLR agonists increased the transcellular permeability of the PPs from Nod2+/+ mice while they did not alter the permeability of Nod2−/− mice (figure 8C). This effect was abrogated when the Nod2+/+ mice were pre-treated with MDP or ML7 (figure 8C). Altogether, these observations showed that TLR stimulation alters the PP permeability through the MLCK pathway and that Nod2 downregulates this effect.

Nod2 downregulates the effect of TLR2/4 agonists on transcellular permeability. (A and B) TLR2 and TLR4 mRNA expressions in PPs from Nod2−/− and Nod2+/+. (C) Transcellular permeability were analysed by FITC–dextran flux. PP from Nod2+/+ and Nod2−/− mice were mounted in Ussing chambers and Pam3CSK4, PGN, LPS, ML-7 or MDP were added into the luminal side. To investigate the roles of Nod2 and MLCK, Nod2+/+ mice were respectively treated with MDP or ML-7 before experiments. Data represent the mean±SEM of eight mice per group. **p <0.01 and ***p <0.001, significantly different from Nod2+/+ under basal condition; §p <0.05, §§p <0.01 and §§§p <0.001, significantly different from Nod2+/+ treated by TLR2 or TLR4 agonist. ABT, antibiotic; FITC, fluorescein isothiocyanate; KO, knock-out; LPS, lipopolysaccharide; MDP, muramyl dipeptide; MLCK, myosin light chain kinase; PGN, peptigoglycan; PPs, Peyer's patches; TLR, toll-like receptor; WT, wild-type.
Previous reports suggest that TLR2/4 activation enhances transepithelial transport of microparticles by M cells and migration of subepithelial dendritic cells (DCs) into the follicle-associated epithelium (FAE).26 Although PPs from Nod2−/− exhibited a similar number of DCs than Nod2+/+ mice,16 we therefore investigated the distribution of DCs in PPs. In vivo, no differences were observed between Nod2+/+ and Nod2−/− mice for the distribution of DCs into the FAE and in the subepithelial compartment of PPs (supplementary figure S4A–D). However, after ex vivo TLR stimulation in Ussing chambers, there was an increased number of DCs into the FAE in comparison with unstimulated PPs as previously reported (supplementary figure S4E,F).
Role of bacterial translocation through Peyer's patches in Nod2 animal models
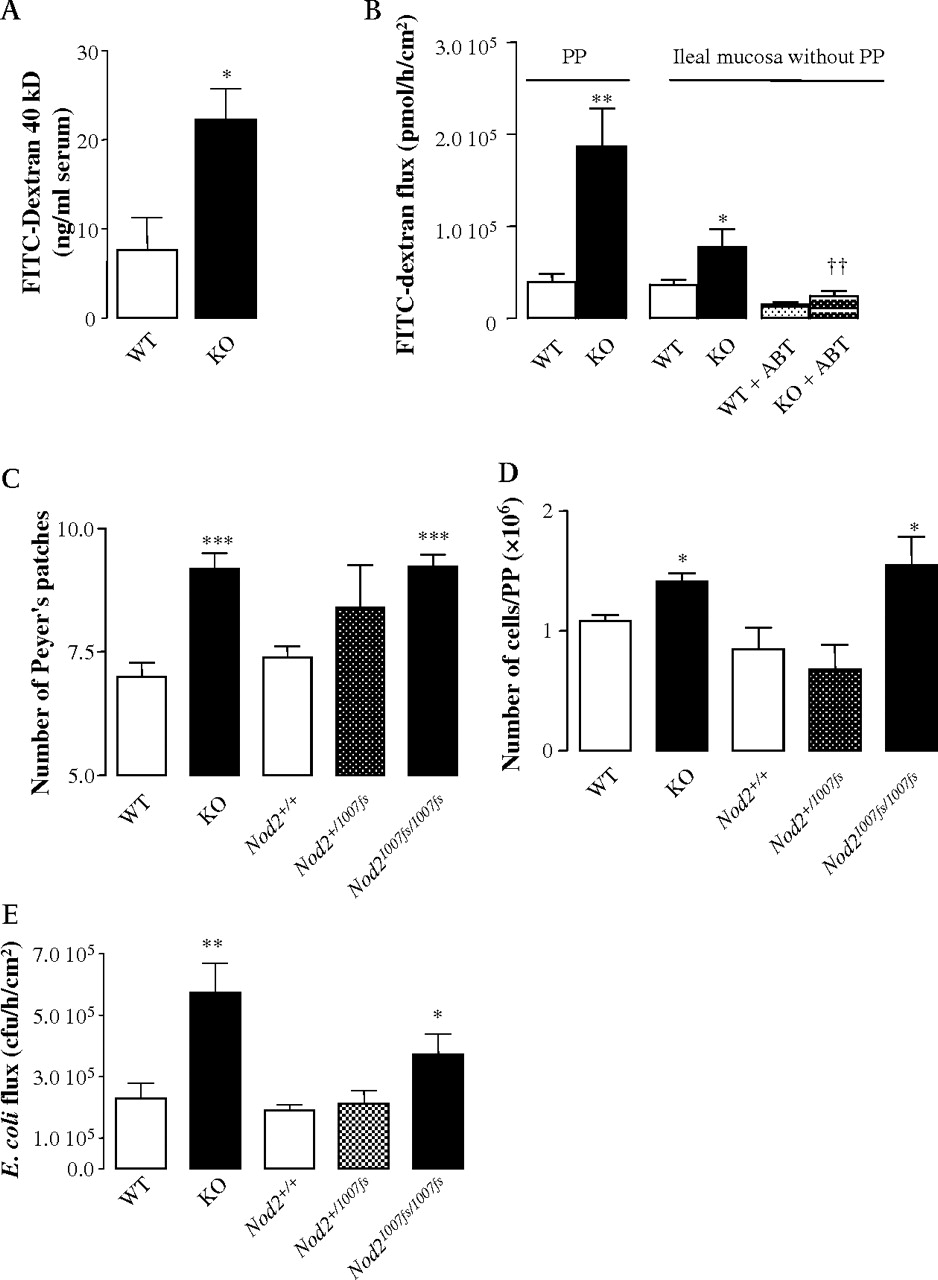
The main phenotype investigated in this work consists in the translocation of bacteria through PP formations. However, the role of the ileal mucosa beside the PP structures is questionable. We have previously shown that out of PP areas, the bacterial translocation of E coli was low and that it was not altered in Nod2−/− mice.16 As a result, we concluded that the role of the ileal mucosa out of LF structures seems to be limited in terms of bacterial translocation. However, it is of note that in the ileum outside the PPs, the transcellular permeability was increased in Nod2−/− mice. This phenotype was reversed by oral ABT (figure 9A). In consequence, it is not possible to rule out a role in antigen transport of the ileum out of the PP areas. In addition, the effect of Nod2 deficiency on the paracellular transport has not been studied and it will require further investigations.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Role of bacterial translocation through PPs in Nod2 animal models. (A) Transcellular permeability was analysed by FITC–dextran 40 kDa flux through PPs and ileal mucosa free of PPs. (B) In vivo transcellular permeability analyses: FITC–dextran 40 kDa was measured in serum from Nod2+/+ and Nod2−/− mice recovered after oral inoculation of the macromolecule. (C) PP counts in the whole intestines of Nod2mut/mut, Nod2snp13/+ and Nod2+/+ mice. (D) Number of immune cells per PP under basal conditions. (E) Bacterial translocation of Escherichia coli through PP formations. Data represent the mean±SEM of at least eight mice per group. *p <0.05, **p <0.01 and ***p <0.001, significantly different from Nod2+/+. FITC, fluorescein isothiocyanate; KO, knock-out; PPs, Peyer's patches; WT, wild-type.
Because our data have been obtained almost exclusively in Ussing chamber experiments, we wanted to confirm the ex vivo increased bacterial translocation in Nod2−/− mice by in vivo methods. For this reason, we used the FITC-labelled dextran 40 kDa (FITC–dextran) as a non-metabolisable transcellular permeability probe. After oral ingestion, the fluorescent particle was recovered at a higher rate in the blood of Nod2−/− mice demonstrating the relevance of our findings in vivo (figure 9B).
Finally, because Crohn's disease and GVHD are associated with Nod2 mutations rather than NOD2 deletion, we looked at the phenotype of the previously described Nod2mut/mut mice carrying a frame-shift mutation homologous to the Crohn's disease associated 1007fs human mutation.27 Like Nod2−/− mice, Nod2mut/mut mice exhibited an increased number of PPs and an increased number of immune cells per PP (figure 9C and D). In addition, Nod2mut/mut mice were also characterised by an elevated translocation of E coli in Ussing chamber experiments (figure 9E).
Discussion
Despite a well-established association between NOD2 mutations and Crohn's disease6 7 as well as GVHD,9 10 the mechanisms by which NOD2 mutations induce these diseases in human are poorly understood. Because Crohn's disease and GVHD have been related to host–microbial interactions and because alterations of PPs have been shown in both diseases,14 15 we focused on these specialised GALT formations. We have previously shown that deletion of Nod2 in mice causes alterations of the structure and the permeability of PP.16 Here we show that these alterations result from an impaired interaction between immune and epithelial cells implicating CD4+ T cell infiltration, IFNγ secretion and MLCK activity in epithelial cells recovering PPs. This PP dysfunction is under the control of the ileal microflora and it may be reproduced by TLR2/4 stimulation. Altogether, these data suggest that Nod2 contributes to the immunogenic tolerance towards gut microflora likely by downregulating the TLR pathways. A schematic representation of the proposed interactions is shown in the supplementary figure S6. Finally, the preliminary data obtained on the mouse model carrying the mutation homologous to the human frame-shift mutation suggest that the proposed model may be relevant for Crohn's disease-associated mutations.
Commensal bacteria play a critical role in the postnatal development and maintenance of the gut-associated lympoid tissue (GALT).28 29 Germ-free animals have an underdeveloped GALT and are resistant to colitis and GVHD. TLRs and NODs have been considered as putative key factors of GALT development under the stimulation the resident flora. TLRs play only a limited role in the postnatal development of PP.30 We have previously reported that Nod2−/− mice exhibit a hyperplasia and a hypertrophy of the GALT after birth, suggesting an important role of Nod2 in the development of GALT during the bacterial colonisation of gut.16 This pivotal role has been herein reinforced by the fact that the Nod2 mutated mice (namely Nod2mut/mut) also exhibit a hyperplasia and a hypertrophy of the PP with a concomitant elevated translocation of E coli. As this now shows that the deletion of the intestinal flora by oral ABT reverses this phenotype, we conclude that Nod2 downregulates the development of the GALT induced by the gut microflora.
It has been shown that the gut microflora drive the expansion of proinflammatory CD4+ T cells in the colonic lamina propria under normal and inflammatory conditions.31 Moreover, Yaguchi et al have shown that treatment of Nod2+/+ mice with by subcutaneous injections of antibiotics decreases the numbers of lymphocytes in PP without significant changes in the lymphocyte phenotype and cytokine levels.32 In agreement with this study, we found that oral ABT reduce the number of T cells in PPs from Nod2+/+ mice without changes in their cytokine contents but, in contrast, we found that oral ABT reduced the percentage of CD4+ T cells. This limited discrepancy could be explained by differences in ABT regimens, our ABT protocol being longer and more efficient to destroy ileal microflora.
In the absence of Nod2, gut microflora exert a stronger stimulation on the PPs as shown by a higher proportion of CD4+ T cells, higher levels of inflammatory cytokines and, finally, higher permeability rates for antigens and bacteria. All of these alterations were suppressed after treatment with oral antibiotics. As a result, Nod2 appears not only to influence the development of the GALT but it is also able to modulate the immune response towards bacteria, by limiting the development of a Th1 immune response. These results are in agreement with data showing that NOD2 activation in DCs regulates their ability to induce a polarised Th1 response in CD4+ T cells.33 In NOD2+/+ DCs, MDP acts synergistically with lipopolysaccharide (LPS) to promote the proliferation of naïve CD4+ T cells with a Th2-like cytokine profile. By contrast, DCs carrying NOD2 mutations are unable to react to MDP, but they respond to LPS and they promote the development of Th1-orientated cells. As a consequence, patients with Crohn's disease who are NOD2 deficient are predisposed to the generation of strongly polarised Th1 response against commensal microorganisms.33
CD4+ T cells play a pivotal role in the pathogenesis of human inflammatory diseases as well as experimental colitis.34 35 Interleukin 2 knock-out (IL2 KO) mice develop spontaneous intestinal inflammation which requires CD4+ T cells.36 The transfer of CD45Rbhigh CD4+ T cell into SCID mice induces an intestinal inflammation.37 Finally, immunotherapy with anti-CD4 may be of benefit in IBD.38 In agreement, we show here that CD4+ depletion fully reverses the phenotype observed in Nod2−/− mice. In Crohn's disease and other intestinal diseases with compromised barrier function, the Th1 immune activation is associated with increased levels of mucosal IFNγ and TNFα. In vitro studies have shown that IFNγ and TNFα induced barrier dysfunction in cultured epithelial monolayers.39 40 Finally, in human and in animal models, IFNγ and TNFα antagonists can diminish disease severity and restore barrier function.23 41–43 In agreement with these observations, our data show that IFNγ over-expression in Nod2−/− mice plays a key role in the disruption of the epithelial barrier integrity of PPs. In addition, it has been shown that IFNγ primes the intestinal epithelium to respond to TNFα by inducing TNFR2 expression, which in turn mediates TNFα-induced MLCK-dependent barrier dysfunction.24 In accordance with these findings, we found that TNFR2 and MLCK mRNA expression are dependent on IFNγ secretion in our model. During the last decade, growing evidence has pointed to the crucial role of MLCK in the barrier dysfunction observed in the pathogenesis of gut inflammation.25 MLCK expression and activity are upregulated in ileal and colonic areas involved in IBD and the degree of upregulation correlates positively with the degree of active inflammation.25 In addition, MLCK is involved in LPS-induced disruption of colonic epithelial barrier and bacterial translocation in rats44 as well as in the barrier dysfunction induced by TNFα.45 46
Here we show that PPs from Nod2−/− mice exhibit an increased expression of the long isoform of MLCK. This isoform, specifically expressed by gut epithelial cells and endothelial cells, is responsible of MLC phosphorylation which modulates the intestinal permeability.47 More importantly, the inhibition of MLCK activity by ML-7 treatment was able to completely reverse the mucosal phenotype in PPs from Nod2−/− mice including CD4+ T cell infiltration and IFNγ and TNFα levels. Altogether, these results demonstrate that MLCK activity plays a crucial role in the maintenance of the increased immune response related with barrier dysfunction.
Recent data from the literature support the opinion that Nod2 mediates an immune tolerance towards bacterial products in human and mouse.2 3 Chronic stimulation of NOD2 results in a decreased production of inflammatory mediators in response to subsequent TLR2/4 stimulation in human macrophages.2 3 This immune tolerance affects the production of TNFα, a cytokine with a central role in the pathogenesis of Crohn's disease.48 NOD2 inhibits the TLR2 pathway by blocking the nuclear translocation of C-Rel.5 This inhibitory effect is mediated by an upregulation of IRF4 (IFN regulatory factor 4).49 NOD2 also inhibits the TLR4 pathway. The inhibition of TLR signalling by NOD2 may be pivotal by preventing an excessive gut inflammation driven by the microflora. We report here an over-expression of TLR2/4 mRNA in PP from Nod2−/− mice. In Nod2+/+ mice, TLR2/4 stimulation triggers increased antigen permeability and migration of DCs into the FAE. This increased permeability is downregulated by MDP pre-treatment, and is dependent on MLCK activity. It can thus be postulated that the phenotype driven by the ileal microflora in Nod2−/− mice is related with the activation of the TLR pathways.
Nod2−/− mice are characterised by an excess of bacterial translocation. This effect can be demonstrated in vivo and ex vivo. It seems to be mainly related to PP areas, bacterial translocation across PP-free ileal mucosa being comparable between Nod2+/+ and Nod2−/− mice. Finally, PP abnormalities are also observed in the mutated mice Nod2mut/mut which carry the mutation homologous with the most comprehensive mutation in humans. This finding suggests it would be useful to further explore the functions of PPs in human diseases, namely Crohn's disease and GVHD.
References
Footnotes
See Editorial, p153
Supplementary figures and tables are published online only at http://gut.bmj.com/content/vol59/issue2
Linked articles 193185
Funding This work was supported by the Institut National de la Santé et de la Recherche Médicale, la Mairie de Paris, BREMICI, Association François Aupetit, Fondation pour la Recherche Médicale and the National Institute of Health.
Competing interests None.
Ethics approval The mice were housed in accordance with the institutional animal healthcare guidelines. The experiments were approved by the institutional committee for animal use.
Provenance and peer review Not commissioned; externally peer reviewed.