Article Text

Abstract
Objective Chronic pancreatitis is a progressive inflammatory disorder of the pancreas characterised by permanent destruction of acinar cells. Mutations in the chymotrypsinogen C (CTRC) gene have been linked to the development of chronic pancreatitis. The aim of the present study was to explore whether CTRC mutants induce endoplasmic reticulum (ER) stress in pancreatic acinar cells.
Design Dexamethasone-differentiated AR42J rat acinar cells and freshly isolated mouse acini were transfected with recombinant adenovirus carrying wild-type CTRC or the p.A73T pancreatitis-associated mutant. ER stress markers were assessed by reverse transcription-PCR and western blotting. Apoptosis was characterised by caspase-3/7 activity and the TUNEL assay.
Results Acinar cells transfected with the p.A73T mutant, but not those with wild-type CTRC, developed significant ER stress as judged by elevated mRNA and protein levels of the ER chaperone immunoglobulin-binding protein (BiP), increased splicing of the X-box binding protein-1 (XBP1) mRNA and marked induction of the transcription factor C/EBP-homologous protein (CHOP), a mediator of ER stress-associated apoptosis. Consistent with higher CHOP expression, AR42J cells expressing the p.A73T mutant became detached over time and showed considerably increased caspase-3/7 activity and TUNEL staining.
Conclusions Pancreatitis-associated CTRC mutations can markedly increase the propensity of chymotrypsinogen C to elicit ER stress in pancreatic acinar cells. Thus, carriers of CTRC mutations may be at a higher risk of developing ER stress in the exocrine pancreas, which may contribute to parenchymal damage through acinar cell apoptosis.
- Chronic pancreatitis
- protein misfolding
- endoplasmic reticulum stress
- unfolded protein response
- apoptosis
- pancreatitis
- trypsinogen
Statistics from Altmetric.com
- Chronic pancreatitis
- protein misfolding
- endoplasmic reticulum stress
- unfolded protein response
- apoptosis
- pancreatitis
- trypsinogen
Chronic pancreatitis is a persistent inflammatory disorder characterised by destruction of the pancreatic parenchyma, maldigestion, chronic pain and diabetes mellitus.1 Identification of genetic risk factors and elucidation of their mechanism of action has allowed the formulation of a disease model in which increased trypsinogen activation and failure of protective mechanisms responsible for trypsin inactivation represent the major pathological pathways.2 3 Four significant milestones contributed to the definition of this trypsin-dependent disease mechanism. First, cationic trypsinogen (PRSS1) mutations were identified in association with hereditary pancreatitis and the majority of these mutations were shown to stimulate autoactivation of trypsinogen to trypsin.2–6 Second, mutations in the SPINK1 gene encoding pancreatic secretory trypsin inhibitor were detected in subjects with idiopathic, alcoholic and tropical pancreatitis.7–9 Many of the SPINK1 mutations studied so far have been found to cause diminished trypsin inhibitor expression at the mRNA and/or protein level, although the main disease-related variant (p.N34S) has yet to be shown to have a functional consequence.10–13 Third, a mutation in anionic trypsinogen (PRSS2) was described which promoted rapid autodegradation and afforded protection against chronic pancreatitis.14 This finding was important conceptually as it highlighted the protective effect of trypsinogen degradation against chronic pancreatitis. Finally, we recently demonstrated that the digestive enzyme chymotrypsin C (CTRC) promoted trypsinogen and trypsin degradation and mutations in the CTRC gene predisposed to chronic pancreatitis.15–17 CTRC mutants exhibited diminished secretion and in some cases also loss of catalytic activity; we therefore proposed that increased risk for chronic pancreatitis in mutation carriers is best explained by the reduced trypsin degrading activity within the pancreas.16
The trypsin-dependent disease model described above assumes that gain or loss of catalytic activity of the participant proteins is critical in disease pathogenesis. We hypothesised, however, that mutations in digestive enzymes might increase the risk of chronic pancreatitis by an alternative mechanism which involves mutation-induced misfolding. Intracellular retention of misfolded proteins results in endoplasmic reticulum (ER) stress and activates a signalling pathway aimed at alleviating the ER burden by increasing protein folding capacity and attenuating translation.18–23 Potentially harmful consequences of this signalling process are the activation of the inflammatory transcription factor NFκB and the induction of apoptotic cell death. In the present study we examined the effect of a representative CTRC mutant, p.A73T, on ER stress markers and apoptosis in the dexamethasone-differentiated rat acinar cell line AR42J and in primary mouse acini.
Methods
Recombinant adenovirus construction
The cDNAs for the human wild-type CTRC and the p.A73T mutant carrying a Glu-Glu epitope tag were excised from the previously constructed pcDNA3.1(-)_CTRC expression plasmids15 16 with XhoI and EcoRI and subcloned into the VQ Ad5CMV shuttle vector under the control of a CMV promoter. Recombinant adenovirus was custom-made by Viraquest Inc (North Liberty, Iowa, USA). Adenovirus containing the enhanced green fluorescent protein (eGFP) cDNA was also purchased from Viraquest. Virus particles were stored in A195 buffer at 1×1012 particles/ml (∼1–4×1010 plaque-forming units (pfu)/ml) concentration at −80°C in aliquots. The infectious titre of the adenovirus stocks was confirmed with the Adeno-X Rapid Titre Kit (Clontech, Mountain View, California, USA).
Cell culture and transfection of AR42J cells
Rat pancreatic AR42J acinar cells were purchased from ATCC (#CRL-1492) and maintained as subconfluent cultures in D-MEM containing 20% fetal bovine serum, 2 mM glutamine and 1% penicillin/streptomycin solution at 37°C in a humidified atmosphere containing 5% carbon dioxide. Prior to transfection, cells were plated into 35 mm wells (106 cells per well) and were grown in the presence of 100 nM dexamethasone (final concentration; #D4902, Sigma-Aldrich, St. Louis, Missouri, USA) for 48 h to induce differentiation.24
Unless indicated otherwise, transfections were performed with 2×108 pfu/ml final adenovirus concentrations in 1 ml OptiMEM supplemented with 2 mM glutamine and 1% penicillin/streptomycin in the presence of dexamethasone (100 nM final concentration).
Preparation and transfection of primary mouse pancreatic acini
Mice were treated according to the Federal Guidelines for Animal Care and the Institutional Animal Care and Use Committee of Boston University approved the animal research protocol. Pancreatic acini were prepared in D-MEM/F-12 medium supplemented with 0.1% BSA, 1 mM sodium pyruvate and 1% penicillin/streptomycin solution (final concentrations). Male Hsd:ICR (CD-1) mice (21–24 g; purchased from Harlan Sprague Dawley Inc) were sacrificed by carbon dioxide inhalation followed by cervical dislocation. The pancreases were excised, digested with collagenase (#C5138, Sigma; 0.25 mg/ml final concentration) at 37°C in an atmosphere containing 100% oxygen, dispersed by pipetting and passed through a 160 μm nylon mesh (Sefar Filtration Inc, Depew, New York, USA, purchased from SmallParts Inc, Miramar, Florida, USA; part# CMN-LP160037-06). Acini were then washed with D-MEM/F-12 medium and plated into 35 mm wells (1 ml aliquots). Before transfection, acinar cells were incubated for 1 h at 37°C in a humidified atmosphere containing 5% carbon dioxide. Transfections were performed with 2×108 pfu/ml final adenovirus concentrations in 1 ml D-MEM/F-12 medium supplemented with 0.1% BSA, 1 mM sodium pyruvate and 1% penicillin/streptomycin solution (final concentrations).
Preparation of cell lysates
Transfected cells were washed twice with phosphate buffered saline (PBS); 200 μl reporter lysis buffer (Promega, Madison, Wisconsin, USA), 4 μl protease inhibitor cocktail (#P8340, Sigma) and 2 μl Halt phosphatase inhibitor cocktail (#78420, Thermo Scientific, Rockford, Illinois, USA) were added and the cells were briefly vortexed. After 15 min incubation at 4°C, the lysates were centrifuged for 5 min at 16 000 g in a microcentrifuge and the pellet was discarded. The protein concentration of the supernatant was measured with the Micro BCA Protein Assay Kit (Thermo Scientific).
Reverse transcriptase (RT)-PCR analysis
RNA was isolated at 24 h after transfection using the RNAqueous kit (Ambion, Austin, Texas, USA) and 1 μg RNA was reverse-transcribed with M-MLV reverse transcriptase (Ambion). Semi-quantitative measurements of cDNA levels for X-box binding protein-1 (XBP1), XBP1s (spliced form), chaperone immunoglobulin-binding protein (BiP) and C/EBP homologous protein CHOP) were performed by PCR using the primers listed in table 1 in the online supplement. As an internal control for mRNA integrity and equal loading, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified using pseudogene-free amplification conditions (see table 1 in online supplement). PCR products were run on 1–2% agarose gels and stained with ethidium bromide. Band intensities were quantitated using Quantity One Software V.4.3.1 (Bio-Rad, Hercules, California, USA).
Western blot analysis
Aliquots of conditioned media (100 μl per lane) or cell lysates (20 μg protein per lane) were electrophoresed on 15% Tris-glycine minigels and transferred onto Immobilon-P membranes (Millipore, Billerica, Massachusetts, USA). For the analysis of BiP expression, 10% gels were used. The membranes were blocked with 5% milk powder solution overnight and incubated with given primary and secondary antibodies for 1 h at room temperature. To detect the Glu-Glu tag, a horseradish peroxidase (HRP)-conjugated goat polyclonal antibody (#ab1267; Abcam, Cambridge, Massachusetts, USA) was used at a dilution of 1:10 000. BiP was detected with a rabbit polyclonal antibody (#ab21685; Abcam) used at a dilution of 1:5000. Phospho‑eIF2α (Ser51) was detected with a rabbit monoclonal antibody (#1090-1; Epitomics, Burlingame, California, USA) used at a dilution of 1:1000. IκBα was detected with a rabbit polyclonal antibody (#9242; Cell Signalling Technology, Beverly, Massachusetts, USA) used at a dilution of 1:2000. Membranes probed with anti-BiP, anti-eIF2α and anti-IκBα primary antibodies were incubated with an HRP-conjugated goat polyclonal anti-rabbit IgG secondary antibody (#31460; Thermo Scientific) at a dilution of 1:20 000. Actin was detected with a mouse monoclonal antibody (#A4700, clone AC-40; Sigma) used at a dilution of 1:1000 followed by HRP-conjugated goat polyclonal anti-mouse IgG (#ab6789; Abcam) at a dilution of 1:2500. HRP was detected using the SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Cell death assays
Cell viability was determined based on the dehydrogenase activity of living cells using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, Maryland, USA). Forty-eight hours after transfection, both detached and attached AR42J cells were collected, washed with PBS and total protein content was determined from an aliquot of the cells using the Micro BCA Protein Assay Kit. Approximately 1×105 cells per well were seeded into a 96-well flat bottom plate in 100 μl PBS. An aliquot (10 μl) of the tetrazolium substrate was added to each well and plates were incubated at 37°C for 2 h, after which the absorbance at 450 nm was measured using a Spectramax Plus 384 microplate reader (Molecular Devices, Sunnyvale, California, USA). Absorbance values were normalised to total protein concentrations.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labelling (TUNEL) detection of DNA fragmentation was carried out using a fluorescein-based detection kit (In Situ Cell Death Detection Kit, Roche Applied Science, Indianapolis, Indiana, USA) according to the manufacturer's instructions. Before transfection, 1×105 AR42J cells per well were seeded into a 96-well flat bottom plate and allowed to grow for 48 h. Transfection was carried out in a final volume of 100 μl and the TUNEL assay was performed 48 h after transfection. Briefly, cultures were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 1 h at room temperature, followed by 2 min incubation with 0.1% Triton X-100 in 0.1% sodium citrate at 4°C to permeabilise the cells. TUNEL reagent (50 μl) was added to air-dried cells and incubated for 60 min at 37°C in a humidified atmosphere in the dark and the samples were analysed under a fluorescence microscope. Cells were washed twice with PBS after each incubation step in this protocol.
Caspase-3/7 activity was measured using the Apo-ONE Homogenous Caspase-3/7 Assay (Promega, Madison, Wisconsin, USA). AR42J cells were scraped and approximately 1×105 cells (in 50 μl PBS) were mixed with the same volume of the Apo-ONE Homogenous Caspase-3/7 reagent in a 96-well black plate. After incubation at room temperature for 2 h, the fluorescence emission was measured at 530 nm, using an excitation wavelength of 485 nm, in a Spectramax Gemini XS fluorescent microplate reader (Molecular Devices). PBS mixed with the same volume of Apo-ONE Homogenous Caspase-3/7 reagent served as a negative control. Caspase activity was corrected for the activity of the negative control and normalised to total protein concentrations.
Fluorescence microscopy
Cells expressing eGFP or subjected to the TUNEL assay were viewed under a Nikon Eclipse TE 300 inverted microscope with a TE-FM epifluorescence attachment.
Statistical analysis
The significance of changes was analysed with the Tukey-Kramer multiple comparisons test; the differences were regarded significant when the p value was <0.05. Results are means±SEM.
Results
Expression of CTRC in pancreatic acinar cells via adenoviral transfection
To investigate whether the p.A73T CTRC mutant causes ER stress in pancreatic acinar cells, we used dexamethasone-differentiated AR42J rat pancreatic acinar cells and freshly prepared primary mouse acini for the transient expression of CTRC. Acinar cells were transfected with recombinant adenoviral vectors carrying wild-type CTRC or the p.A73T mutant. As a negative control, an adenovirus carrying the eGFP gene was used. The efficiency of transfection exceeded 90% both in AR42J cells (figure 1A) and in primary mouse acini (data not shown). Transfection of AR42J cells with increasing concentrations of recombinant CTRC adenovirus resulted in secretion of CTRC into the growth medium as judged by SDS-PAGE analysis (figure 1B). Secreted levels of CTRC increased as a function of the virus concentration and a robust CTRC band was detected on gels at 108 pfu/ml and above. Primary mouse acini transfected with CTRC adenovirus secreted CTRC protein which was readily detectable by western blots but could not be visualised on Coomassie-stained SDS-PAGE gels due to the complex mixture of secretory proteins produced by the acini and the presence of high concentrations of bovine serum albumin (the Coomassie-stained gels were reviewed but are not shown). It is also important to note that, in AR42J cells and mouse acini, CTRC is expressed in its inactive zymogen form and we detected no signs of spontaneous activation either in the conditioned medium or in cell lysates.

Transfection of AR42J cells with enhanced green fluorescent protein (eGFP) and chymotrypsinogen C (CTRC) adenovirus. (A) Efficiency of transfection as monitored by eGFP expression. Cells were analysed under a fluorescence microscope 24 h after infection (200-fold magnification, scale bars 50 μm): left panel, phase-contrast image; right panel, fluorescent image. (B) CTRC secretion by AR42J cells transfected with increasing concentrations of CTRC adenovirus (expressed in plaque-forming units per ml). Twenty-four hours post-transfection 100 μl aliquots of conditioned media were precipitated with 10% trichloroacetic acid, heat denaturated at 95°C for 5 min in reducing Laemmli sample buffer and electrophoresed on 15% SDS-polyacrylamide gels. Gels were stained with Coomassie Blue.
Diminished secretion and intracellular retention/degradation of the p.A73T CTRC mutant
We first compared spontaneous (basal) and secretagogue (cerulein)-stimulated secretion of wild-type CTRC and the p.A73T CTRC mutant from transfected AR42J cells and mouse acini. Secretion of the p.A73T mutant was markedly reduced compared with wild-type CTRC in both cell types (figure 2A). In AR42J cells the p.A73T mutant was detected in cell lysates at levels that were comparable or higher than those of wild-type CTRC, indicating that the p.A73T mutant is synthesised normally inside the cell but undergoes degradation instead of secretion (figure 2B). In lysates of mouse acini the p.A73T mutant was found in significantly lower amounts than wild-type CTRC, suggesting that the mutant becomes degraded more efficiently in this cell type (figure 2B).

Diminished secretion and intracellular retention/degradation of the p.A73T chymotrypsinogen C (CTRC) mutant. Western blot analysis of (A) conditioned media and (B) cell lysates of pancreatic acinar cells expressing wild-type CTRC, enhanced green fluorescent protein (control) or the p.A73T CTRC mutant. Twenty-four hours after transfection cells were washed twice with phosphate buffered saline and fresh OptiMEM medium was added. After 15 min incubation at 37°C, the medium was collected (‘basal’) and fresh OptiMEM containing 10 nM (AR42J) or 100 pM (mouse acini) concentration of cerulein was added to stimulate secretion. The ‘cerulein’ medium was collected after 15 min incubation at 37°C and cells were also harvested for western blot analysis. Aliquots of conditioned media (100 μl) and cell lysates (20 μg total protein) were loaded onto a 15% SDS-polyacrylamide gel, transferred to Immobilon-P membrane and CTRC was detected with an antibody against the Glu-Glu epitope tag. Representative blots of three independent experiments are shown.
ER stress in AR42J cells expressing the p.A73T CTRC mutant
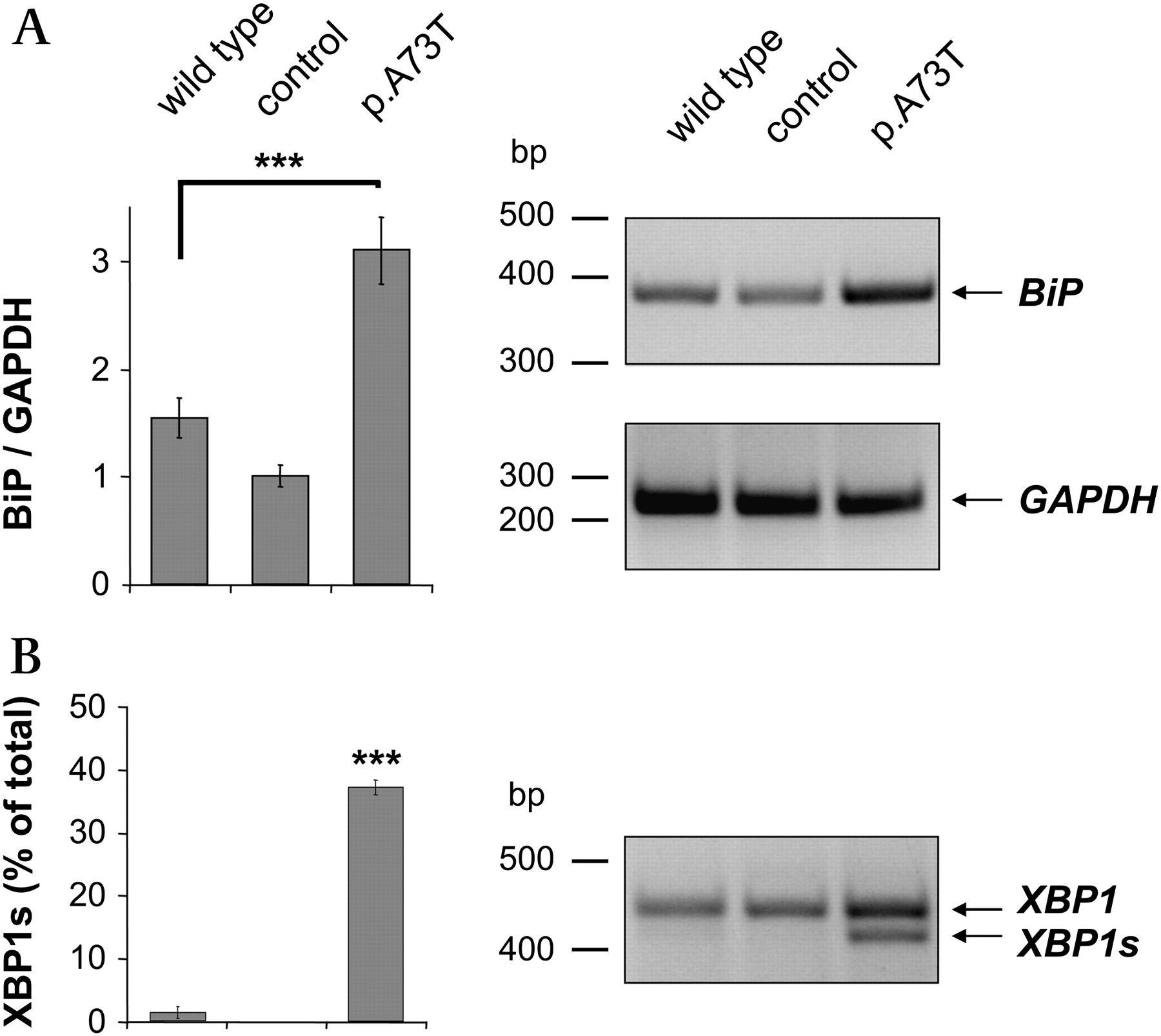
Intracellular retention of the p.A73T mutant most likely occurs in the ER due to mutation-induced misfolding which then results in degradation through the ER-associated protein degradation (ERAD) pathway. If this were the case, the p.A73T mutant might cause ER stress and trigger the unfolded protein response, a signal transduction pathway aimed at alleviating ER protein burden and increasing ER folding capacity. The master regulator of the unfolded protein response is BiP, which becomes characteristically upregulated under conditions of ER stress. To test whether expression of the p.A73T mutant in AR42J cells causes ER stress, we therefore measured the protein and mRNA levels of BiP in cell lysates by western blotting (figure 3A) and semi-quantitative RT-PCR analysis (figure 3B). We found that BiP was significantly upregulated in acinar cells expressing the p.A73T mutant relative to cells transfected with wild-type CTRC or control adenovirus.

Endoplasmic reticulum stress markers in AR42J cells expressing wild-type chymotrypsinogen C (CTRC), enhanced green fluorescent protein (control) or the p.A73T CTRC mutant. (A) Levels of chaperone immunoglobulin-binding protein (BiP) protein (78 kDa) in cell lysates were analysed by western blotting as described in the Methods section. Actin (42 kDa) was measured as a loading control. Band intensities were quantitated by densitometry and BiP/actin ratios are shown as bar graphs. (B) Semiquantitative RT-PCR was performed to determine BiP (578 bp) mRNA levels. Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (261 bp) was measured as a reference control. Band intensities were quantitated by densitometry and BiP/GAPDH ratios are depicted as bar graphs. (C) The extent of X-box binding protein-1 (XBP1) splicing was determined by measuring the unspliced (XBP1; 447 bp) and spliced (XBP1s; 421 bp) forms by RT-PCR. Band intensities were quantitated by densitometry and the amount of the spliced form is shown as percentage of total XBP1 (unspliced and spliced). Values are means±SEM. ***p<0.001, n=3.
In non-stressed mammalian cells, BiP binds to the luminal domain of the membrane-embedded inositol-requiring enzyme 1 (IRE1) and keeps this ER stress sensor protein in an inactive state. During ER stress, BiP dissociates from IRE1 and binds to misfolded proteins, thereby allowing the dimerisation and activation of IRE1. IRE1 catalyses the cytoplasmic splicing of the X-box binding protein-1 (XBP1) mRNA by removing a 26-nucleotide intron to create a shorter variant (XBP1s) which encodes a potent transcriptional activator of the unfolded protein response. We used RT-PCR to measure the extent of XBP1 splicing in AR42J cells expressing the p.A73T mutant. Figure 3C shows that expression of the p.A73T mutant caused a significant increase in XBP1s compared with the lower levels observed in cells expressing wild-type CTRC or infected with control virus.
ER stress in AR42J cells is proportional to intracellular levels of the p.A73T CTRC mutant
To demonstrate that ER stress is dependent on the intracellular accumulation of the misfolded p.A73T CTRC mutant, we measured the time course of CTRC expression for both wild-type and mutant CTRC and compared it with the time course of XBP1 splicing. As shown in figure 4, expression of CTRC in cell lysates became clearly detectable at 8 h, increased by 12 h and changed very little between 12 and 24 h. XBP1 splicing in cells expressing the p.A73T mutant showed very similar kinetics. Thus, minimal splicing was detected already at 4 h, splicing became robust at 8 h and increased to near completion at 12 h. Although total XBP1 levels were somewhat higher at 24 h, the extent of splicing did not change significantly between 12 h and 24 h. In stark contrast to the p.A73T CTRC mutant, cells expressing wild-type CTRC showed only negligible XBP1 splicing by 8 h which remained unchanged at 12 h and 24 h, despite the higher intracellular CTRC content at the later time points. These data clearly demonstrate a positive correlation between ER stress and the intracellular levels of the p.A73T CTRC mutant. Importantly, the results also confirm that heterologous overexpression of CTRC alone is insufficient to trigger significant ER stress.

Time course of chymotrypsinogen C (CTRC) expression and X-box binding protein-1 (XBP1) splicing in AR42J cells expressing (A) wild-type CTRC or (B) the p.A73T CTRC mutant. Control cells were infected with enhanced green fluorescent protein (eGFP) adenovirus and harvested at 24 h. Levels of CTRC protein in cell lysates were analysed by western blotting as described in figure 2 and the Methods section. XBP1 splicing was measured by RT-PCR as described in figure 3 and the Methods section. Expression of glyceraldehyde-3-phosphate dehydrogenase GAPDH) (261 bp) was measured as a control for mRNA integrity and equal loading.
ER stress in primary mouse acinar cells expressing the p.A73T CTRC mutant
We determined mRNA levels for BiP and XBP1s in freshly prepared mouse pancreatic acini using RT-PCR (figure 5). Both ER stress markers were significantly elevated in acini expressing the p.A73T mutant compared with cells infected with wild-type CTRC virus or control virus. We were unable to evaluate intracellular BiP protein levels in a reliable manner as mouse acini exhibited robust BiP secretion which resulted in variable BiP levels in cell lysates. The unusual phenomenon of BiP secretion by pancreatic acini has been reported previously, although its mechanism and physiological significance is unclear.25

Endoplasmic reticulum stress markers in mouse pancreatic acinar cells expressing wild-type chymotrypsinogen C (CTRC), enhanced green fluorescent protein (control) or the p.A73T CTRC mutant. (A) Semiquantitative RT-PCR was performed to determine chaperone immunoglobulin-binding protein (BiP) (398 bp) mRNA levels. Expression of glyceraldehyde-3-phosphate dehydrogenase GAPDH) (261 bp) was measured as a reference control. Band intensities were quantitated by densitometry and BiP/GAPDH ratios are depicted as bar graphs. (B) The extent of X-box binding protein-1 (XBP1) splicing was determined by measuring the unspliced (XBP1; 447 bp) and spliced (XBP1s; 421 bp) forms by RT-PCR. Band intensities were quantitated by densitometry and the amount of the spliced form is shown as the percentage of total XBP1 (unspliced and spliced). Values are means±SEM. ***p<0.001, n=3.
Activation of the PERK pathway and NFκB is not observed in acinar cells expressing the p.A73T CTRC mutant
Translation attenuation in response to ER stress is mediated by the protein kinase RNA (PKR)-like endoplasmic reticulum kinase (PERK), an ER membrane protein. Dimerisation of PERK leads to trans-autophosphorylation and activation of its cytoplasmic protein kinase domain, which in turn phosphorylates the α subunit of the eukaryotic translation initiation factor-2 (eIF2α) thereby impeding translation initiation. One of the consequences of translation attenuation is the activation of NFκB owing to the relative depletion of its short half-life inhibitor IκBα. To assess whether expression of the p.A73T mutant causes translation attenuation and NFκB activation, we measured levels of phosphorylated eIF2α and IκBα in lysates of AR42J and mouse pancreatic acinar cells at 24 h after transfection by western blot analysis (see figure 1 in online supplement). No significant changes were detected in these markers in cells expressing the p.A73T mutant compared with cells transfected with wild-type CTRC or control virus, indicating that the PERK pathway was not activated. To assess whether translation attenuation might be more prominent at earlier time points, we followed the time course of eIF2α phosphorylation in AR42J cells expressing the p.A73T CTRC mutant but found no increase in phosphorylation at 4, 8, and 12 h (reviewed but not shown).
Apoptotic cell death in AR42J cells expressing the p.A73T CTRC mutant
Chronic ER stress may trigger apoptosis and we observed that AR42J cells expressing the p.A73T mutant became almost completely detached from the tissue culture plates within 48 h, whereas cells expressing the wild-type CTRC or those infected with control virus remained attached to the plastic support and showed normal morphology (figure 6A). The viability of the acinar cells expressing the p.A73T CTRC mutant was also significantly decreased when assessed by an assay based on the dehydrogenase activity of living cells (figure 6B). To obtain evidence that AR42J cells expressing the p.A73T mutant undergo apoptosis, we measured caspase 3/7 activity from cell lysates at 24 h and 48 h after transfection. Caspase activity increased between 24 h and 48 h by 2–3-fold, and at both time points it was significantly higher in cells expressing the p.A73T mutant than in cells transfected with wild-type CTRC or control virus (figure 7A). Furthermore, at 48 h after transfection, TUNEL staining revealed a striking increase in the number of apoptotic nuclei in cells expressing the p.A73T mutant compared with cells expressing wild-type CTRC (figure 7B).

Viability of AR42J cells expressing the p.A73T chymotrypsinogen C (CTRC) mutant. (A) Detachment of cells from the tissue culture plates. Cells expressing wild-type CTRC or the p.A73T CTRC mutant were co-transfected with enhanced green fluorescent protein (eGFP) for better visualisation. Control cells were transfected with eGFP adenovirus only. At the indicated times, cells were washed twice with phosphate-buffered saline to remove detached cells and photographed under a fluorescence microscope (100-fold magnification, scale bars 100 μm). (B) The number of viable cells expressing wild-type CTRC (WT) or the p.A73T mutant CTRC (M) was estimated 48 h after transfection by their dehydrogenase activity as described in the Methods section. In this assay, both the detached and attached cells were included. Cell viability was expressed as the percentage of the number of living cells in non-transfected control (C) samples. Values are means±SEM. ***p<0.001, n=3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Endoplasmic reticulum stress-induced apoptosis in AR42J cells expressing the p.A73T chymotrypsinogen C (CTRC) mutant. (A) Caspase 3/7 activity was measured from cells expressing wild-type CTRC (WT) or the p.A73T CTRC mutant (M) at 24 h and 48 h after transfection, as described in the Methods section. Caspase activities were expressed as the percentage of the activity measured in the non-transfected control (C) cells at 24 h. (B) TUNEL assay. Cells expressing wild-type CTRC or the p.A73T mutant were fixed at 48 h after transfection and apoptotic nuclei were detected by TUNEL staining. Upper panels show phase contrast images of cells and lower panels show TUNEL-positive apoptotic cells, which were seen in green colour under the fluorescence microscope. Representative photographs are shown at 100-fold magnification (scale bars 100 μm). The bar graphs present the number of TUNEL positive cells counted from six randomly chosen 1 mm2 fields in each well (means±SEM). (C) Semiquantitative RT-PCR was performed to determine C/EBP-homologous protein (CHOP) (286 bp) mRNA levels. The expression of glyceraldehyde-3-phosphate dehydrogenase GAPDH) (261 bp) was measured as a reference control. Band intensities were quantitated by densitometry and CHOP/GAPDH ratios are depicted in bar graphs. Values are means±SEM from three independent experiments (***p<0.001).
ER stress-associated apoptosis is induced by the transcription factor C/EBP homologous protein (CHOP) which becomes highly expressed in ER stress and downregulates expression of anti-apoptotic proteins. We measured the expression of CHOP mRNA in AR42J cells at 24 h after transfection by RT-PCR and found that CHOP levels were significantly elevated in cells expressing the p.A73T mutant relative to cells transfected with wild-type CTRC or control virus (figure 7C). Elevated CHOP levels were also found in primary mouse acini expressing the p.A73T mutant at 24 h, indicating that activation of pro-apoptotic pathways by ER stress has occurred (see figure 2 in online supplement). However, we did not observe signs of increased apoptosis at this early time point, as assessed by viability assays, caspase activity measurements or TUNEL staining (not shown). We were unable to evaluate apoptosis at longer culture times as the viability of primary cultures of acini kept beyond 24 h progressively deteriorated.
Discussion
In the present study we demonstrate that a pancreatitis-associated mutation can increase the ability of CTRC to cause ER stress and subsequent cell death by a mechanism which is unrelated to the trypsin-degrading activity of CTRC but involves mutation-induced misfolding. Previous studies on the mechanism of genetic risk in chronic pancreatitis led to the formulation of a trypsin-dependent disease model which posits that sustained intrapancreatic conversion of trypsinogen to trypsin has a central role in the development of chronic pancreatitis.2–17 One of the defence mechanisms against unwanted trypsin activity in the pancreas is CTRC, which was recently shown to degrade trypsin and trypsinogen with high specificity.15 Mutations in CTRC that abolish catalytic activity and/or decrease secretion have been found in association with chronic pancreatitis, suggesting that failure of the intrapancreatic trypsin-degrading mechanisms increases the risk of pancreatitis.16 17 To date, three CTRC mutations have been confirmed statistically to increase the risk of chronic pancreatitis. Mutations p.R254W and p.K247_R254del are prevalent in European subjects whereas mutation p.A73T was found with higher frequency in India. When analysed in transiently transfected HEK 293T cells, all three mutants exhibited a secretion defect apparently as a result of intracellular retention and degradation.16 Secretion of mutant p.R254W was reduced by about 50%, whereas mutants p.K247_R254del and p.A73T were secreted only in trace amounts. This common phenotype led us to speculate that intracellular retention and degradation of CTRC mutants might cause ER stress which can contribute to the development of chronic pancreatitis. This would represent a novel disease mechanism unrelated to the trypsin-degrading activity of CTRC and thus unrelated to intrapancreatic trypsin levels. We note, however, that loss of function mutations in CTRC might result in ER stress by other mechanisms as well, which might involve accumulation of trypsin or other malformed proteins which would be otherwise degraded by CTRC.
In the present study we established an adenovirus-mediated transfection system for the expression of CTRC in pancreatic acinar cells. Using dexamethasone-differentiated AR42J pancreatic acinar cells and freshly isolated mouse acini, we found that mutant p.A73T was intracellularly retained and degraded, and markers of ER stress were significantly elevated in cells expressing the p.A73T CTRC mutant relative to cells transfected with wild-type CTRC or a control adenovirus. Furthermore, we observed that AR42J cells underwent apoptotic cell death as a result of expressing the p.A73T CTRC mutant. Apoptosis was related to ER stress, as evidenced by marked induction of the pro-apoptotic transcription factor CHOP. We also measured elevated levels of CHOP in primary mouse acinar cells expressing the p.A73T mutant; however, we could not detect apoptosis in these cells during short-term culturing. Mouse acinar cells have been reported to exhibit higher resistance to apoptosis than rat acinar cells, which is consistent with our findings.26
We have previously observed that the hereditary pancreatitis-associated p.R116C cationic trypsinogen mutant suffered intracellular misfolding and retention and caused ER stress in transiently transfected HEK 293T cells.27 Taken together, the two studies propose a new paradigm for the mechanism of genetic risk in chronic pancreatitis which involves ER stress caused by mutation-induced misfolding and ER retention of digestive (pro)enzymes or other secretory proteins. This model implies that the physiological function or catalytic activity of the mutant protein is immaterial, and that mutations in a variety of secretory proteins can increase the risk of pancreatitis as long as the protein is highly expressed and the mutation causes misfolding. ER stress-induced apoptosis can accelerate the loss of functional acini and thus contribute to exocrine insufficiency, a hallmark of chronic pancreatitis. Apoptosis of pancreatic acinar cells was shown to be protective against necrosis and severe inflammation in acute models of experimental pancreatitis,26 but in a chronic setting apoptosis would be clearly detrimental with respect to parenchymal cell loss. This notion is also supported by a recent study which showed that, in the pancreatic tissue of patients with chronic pancreatitis, the number of apoptotic acinar cells was increased 10-fold compared with cells from normal pancreases.28
Summary box
Loss of function mutations in the chymotrypsinogen C (CTRC) gene have been linked to the development of chronic pancreatitis.
Increased pancreatitis risk has so far been explained by the loss of trypsin-degrading activity.
New findings indicate that CTRC mutations can also increase the propensity of chymotrypsinogen C to elicit endoplasmic reticulum (ER) stress in pancreatic acinar cells.
Carriers of CTRC mutations may be at a higher risk of developing ER stress in the exocrine pancreas.
ER stress may contribute to parenchymal damage in chronic pancreatitis through acinar cell apoptosis.
Acknowledgments
The authors thank Rajinder Dawra and George Perides for helpful technical suggestions.
References
Supplementary materials
Web only data 59:3;365
Files in this Data Supplement:
Footnotes
Funding This work was supported by US National Institutes of Health grants DK082412 and DK058088 (to MS-T) and a grant from the National Pancreas Foundation (to RS).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.