Article Text

Statistics from Altmetric.com
Introduction
A stem cell niche is the restricted compartment in a tissue that maintains and regulates stem cell behaviour, supporting self-renewal and maintaining the balance between quiescence, proliferation and differentiation required in response to injury.1 2 The existence of a niche structure was first proposed for haematopoietic stem cells in the bone marrow (BM)3 and in gonads in invertebrate models.4 5 In humans, the intestinal mucosa crypt has been extensively studied as a model of adult stem cell niche.6
In 1958, Wilson and Leduc7 described a cell population in the distal biliary ducts of the liver capable of both hepatocyte and cholangiocyte differentiation. Subsequent studies8 9 suggested that hepatocytes and bile duct epithelial cells were of common embryonic origin deriving from a common bipotential progenitor. The canals of Hering, the terminal branches of the intrahepatic biliary system, have been proposed as the source of those bipotential liver cells, termed oval cells (OCs) in rodents and hepatic progenitor cells (HPCs) in humans.10 OCs are widely considered to be putative liver stem cells that can regenerate the parenchyma when hepatocyte proliferation is overwhelmed by persistent or severe liver injury. Recent studies have also suggested that deregulated OCs might be a potential source of liver cancer (eg, hepatocellular carcinoma and cholangiocarcinoma).11
Non-parenchymal cells (NPCs) in the liver include stellate cells/myofibroblasts, which are the main producers of collagen; macrophages, which are involved in tissue remodelling and fibrosis resolution after extensive damage12; endothelial cells, which are able to form new vessels13; and other leucocytes recruited by local inflammation. NPCs produce cytokines and growth factors, like transforming growth factor β, that influence OC/HPC and hepatocyte proliferation,14 but most of the signals they exchange with the OC/HPC compartment and their role in regulating OC/HPC behaviour has yet to be fully elucidated.15 Moreover, studies have demonstrated that in liver injury a proportion of myofibroblasts and macrophages are recruited from the BM.16 17 It has been claimed that OCs are of BM origin18 19; however, other studies have found that OCs are intrinsic to the liver and not of BM origin.20 We have used a dietary means of OC induction in BM transplanted mice to track which cells within the niche are of BM origin.
Cell–cell interaction and also cell–matrix interplay are likely to be important in regulating stem cell behaviour within niches.2 In the liver, the extracellular matrix and basement membrane of the bile ducts, where OCs/HPCs reside, is mainly composed of laminin and type IV collagen.21 22 Interestingly, laminin gene expression has been documented in NPCs in the liver and, in particular, in hepatic stellate cells and endothelial cells.23 24 In the 2-acetylaminofluorene model of liver injury in rats, a laminin-rich basement membrane has been shown to be intimately associated with the OC response.25 However, whether this is a general phenomenon in liver progenitor activation and the functional significance of the laminin matrix–progenitor cell interaction is not known.
To determine whether a stereotypical OC/HPC niche forms during liver regeneration, we deliberately analysed the liver tissue from a wide variety of liver injury models in rodent and human tissue and compared it with undamaged tissue. Having determined that a laminin matrix always surrounds the OC/HPC response, we studied the functional consequences of the laminin-rich basement membrane upon OC behaviour. Culturing OCs on various matrices we demonstrated that laminin allows maintenance of OCs in a progenitor/biliary phenotype, and inhibits hepatocyte differentiation. This is consistent with the hypothesis that the adult liver progenitor cell niche is a specialised environment where progenitor cells can remain undifferentiated and proliferate in response to injury, and upon leaving the laminin niche the cells differentiate into a hepatocyte phenotype.
Although progenitor cells are important for regenerating the liver in severe damage, deregulated OC/HPC proliferation might be a source of liver cancer.11 26 27 Understanding how the niche influences OC/HPC behaviour is therefore likely to be of scientific and clinical importance if novel strategies are to be developed that can promote liver regeneration in the setting of chronic liver injury. Finally, while it is now accepted that niches are important for controlling stem/progenitor cell behaviour in several stem cell systems, the progenitor cells themselves may also influence the niche environment, including that of the liver.28 Clearly, this could be important in diseases such as fibrosing cholestatic hepatitis where very rapid fibrosis is characteristic and ductular reactions/HPC responses are seen.
Material and methods
Animal models
All animal work was carried out under procedural and ethical guidelines of the Home Office (UK). Three independent rodent models of OC activation were used to assess the cellular constituents of the liver tissue niche: (a) Male Fischer rats weighing 200 g were treated with 2-acetylaminofluorene and subsequent partial hepatectomy as previously described.29 Livers were analysed from 3 to 13 days after partial hepatectomy. Livers from normal rats were used as controls. (2) Twenty-six-week-old female mice, transgenic for the hepatitis B surface antigen (HBsAg) were used. These mice have hepatocyte-specific expression of the hepatitis B virus BgIII-A fragment and develop hepatocellular injury. To induce OC proliferation animals were treated with the pyrrolizidine alkaloid, retrorsine, (70 mg/kg intraperitoneally, Sigma-Aldrich, Dorset, UK) with a further injection 2 weeks later. Liver tissue was analysed 6 months later. Liver from age-matched C57/B6 animals was used as control. (c) In a dietary protocol of OC induction C57/B6 mice were fed a diet composed of powdered choline-deficient chow (MP Biomedicals, Cambridge, UK) mixed in a 1:1 ratio with normal powdered chow (called the 50% CDE diet) supplemented with dl-ethionine (Sigma) at 0.15% in sweetened water (to improve animal tolerance to ethionine). To enable BM tracking, 6-week-old female mice were irradiated with whole-body gamma irradiation (10.5 Gray) followed by injection with 1×107 BM cells extracted from 6-week-old male femurs. Mice were allowed to reconstitute for 6 weeks and then started the diet or at 2 weeks before tissue analysis. Mice were treated for 1 week before and for 4 weeks after BM transplantation with Baytril antibiotics (W&J Dunlops, Dumfries, UK).
Human liver tissue
The human tissue analysed belonged to one of the following categories: (a) control liver tissue obtained from a pre-perfusion liver biopsy specimen (n=1); (b) tissue from patients with hepatitis C virus-related chronic hepatitis (n=10); (c) tissue from patients with hepatitis B virus chronic hepatitis (n=10); (d) tissue obtained from biopsies performed from 0 to 6 months after liver transplant in a patient who received a liver transplant for hepatitis C and developed recurrent fibrosing cholestatic hepatitis after transplant (n=1).
Immunohistochemistry (IHC)
Paraffin-embedded sections (3–5 μm) were dewaxed and rehydrated before IHC, which was performed using a standard avidin/biotin method. OCs (rodent tissue) and HPCs (human tissue) were identified using antibodies against either cytokeratin 19 (rat tissue; dilution 1:50, Novocastra, Newcastle-upon-Tyne, UK), wide-spectrum cytokeratin (mouse tissue; dilution 1:200, Dako, Cambridge, UK) or cytokeratin 7 (human tissue; dilution 1:100, Dako).
ED1 (rat tissue; dilution 1:400, Serotec), F4/80 (mouse tissue; dilution 1:10, eBioscience, Hatfield, UK) or CD68 (human tissue, dilution 1:200, Dako) were used to identify macrophages. Myofibroblasts were immunostained using an antibody to α smooth muscle actin (αSMA) (dilution 1:4000 (or 1:2000 for immunofluorescence), Sigma). Endothelial cells were detected using a von Willebrand factor (vWF) antibody (dilution 1:50, Dako). Laminin was detected using a rabbit polyclonal antibody to laminin (dilution 1:25, Dako). Clone numbers of primary antibodies are specified in table 1. For immunofluorescence goat anti-rabbit Alexa-488 (Invitrogen, Paisley, UK) conjugated, streptavidin Alexa-555 (Invitrogen) conjugated goat anti-mouse Cy5-labelled (Abcam, Cambridge, UK) secondary antibodies were used.
Clone numbers of primary antibodies
Fluorescent in situ hybridisation (FISH)
After IHC, sections were washed in phosphate-buffered saline (PBS) and incubated in 1 M sodium thiocyanate in distilled water for 10 min at 80°C, washed in PBS and digested in 0.4% w/v pepsin in 0.1 M HCl at 37°C for 4 min, quenched in 0.2% glycine in double concentration PBS for 2 min, post-fixed in 4% paraformaldehyde for 2 min, dehydrated through graded alcohols and air dried. A fluorescein isothiocyanate-labelled Y chromosome paint (Star-FISH, Cambio, Cambridge, UK) was added to sections, sealed under a glass cover slip and heated to 60°C for 10 min before overnight incubation in a 37°C water bath. Slides were then washed in formamide (50% w/v)/2× saline sodium citrate (SSC) at 37°C, then with 2× SSC and, finally, with 4× SSC/Tween-20 (0.05% w/v) at 37°C. The slides were rinsed in 0.5× SSC at 37°C, then PBS before being mounted with 4′,6-diamidino-2-phenylindole containing mounting media (Vector Laboratories, Peterborough, UK).
Oval cell induction, progenitor cell isolation, culture and analysis
Eight-week-old C57/Bl6 mice were fed the CDE diet for 12 days, excised livers were minced and digested using 200 ng/ml DNAse1 (Roche, Hertfordshire, UK) and 1.25 mg/ml (=125 collagen-digesting units/mg solid) collagenase B (Sigma) in Leibovitz-15 media (Gibco, Paisley, UK). Digested liver was passed through a 40 μm cell strainer (BD Falcon, BD Biosciences, Oxford, UK) and the resulting suspension was spun over a 20%/50% discontinuous Percoll gradient (Sigma). Cells isolated from the 20%/50% boundary were resuspended in BD iMag buffer (BD Biosciences) and labelled with anti-CD45R beads (BD Biosciences), according to the manufacturer's instructions. Labelled cells were separated from other cells using a BD magnet. Those cells remaining after magnetic separation were used in culture. Cells were plated on plastic at a density of 3×10−4 cells/cm2 and cultured for 7 days in Dulbecco's modified Eagle's medium (PAA Laboratories, Yeovil, UK) and Hams-F10 (Gibco). This was supplemented with 10% fetal calf serum (50 ng/ml insulin (Sigma) and 20 ng/ml hydrocortisone (Sigma). Sodium Pyruvate (PAA Laboratories), l-glutamine (PAA) and gentamicin (Gibco). Cells were then trypsinised and plated on 12-well tissue culture plates at 1×104 cells/cm2 coated with laminin, collagen I, collagen IV or fibronectin, according to the manufacturer's instructions (all Sigma) with culture medium as before. The following day culture medium was supplemented with 5% fetal calf serum and 50 ng/ml epidermal growth factor (Sigma) and left for 5 days to differentiate, with media and epidermal growth factor being replaced every 2 days.
For gene expression analysis total mRNA was isolated from cells using the Qiagen RNeasy Mini kit (Qiagen, Crawley, UK) and reverse transcribed using the Quantitect Reverse Transcription kit (Qiagen), according to the manufacturer's instructions. Gene expression analysis was conduced using an Applied Biosystems 7500 thermocycler (Applied Biosystems, Warrington, UK), QuantiFast SYBR assay (Qiagen) and QuantiTect (Qiagen). All samples were run in triplicate. Statistical analysis using a two-tailed Student t test was performed using Prism software (GraphPad Software).
Results
Composition of the OC/HPC niche
Consistent with previous reports, our models of rodent liver injury and injured human liver displayed a prominent expansion of the cytokeratin-positive OC/HPC compartment compared with control tissue (online supplementary figure 1).
Only diffusely spread macrophages (black arrows, brown) were seen in uninjured rodents and human liver and no clear spatial proximity to the bile ducts was discernible (figure 1A–C). In injured liver tissue (figure 1D–F), macrophages were found both clustered in areas where the OC/HPC reaction was evident, and distributed in tissue outside the site of the progenitor cell response. Close contact between macrophages and OCs/HPCs can be seen in double-stained images.

Macrophages. Photomicrographs illustrating rodent and human liver tissue to visualise the partner cells of the hepatic stem cell niche. In control liver tissue (A–C) the macrophage-specific staining shows only a few macrophages (black arrows) scattered near the portal areas but without any specific relationship with the bile ducts. (A) Normal rat liver, double staining for ED1 (brown) and CK19 (red); (B) normal mouse liver, F4/80 (brown); (C) biopsy specimen from control pre-perfusion donor liver, CD68 (brown). In damaged liver tissue (D–F), macrophages (black arrows) increase in number around the oval cells (OCs)/hepatic progenitor cells. (D) 2-Acetylaminofluorene and subsequent partial hepatectomy (2-AAF/PH) rat, day 9 after the PH—double staining for cytokeratin 19 (red) and ED1 (brown); (E) hepatitis B surface antigen-tg/retrorsine-treated mouse—F4/80 (brown); (F) hepatitis C virus-related cirrhotic human liver—double staining for CK7 (red) and CD68 (brown). Magnification (A–E) ×40; (F) ×60.
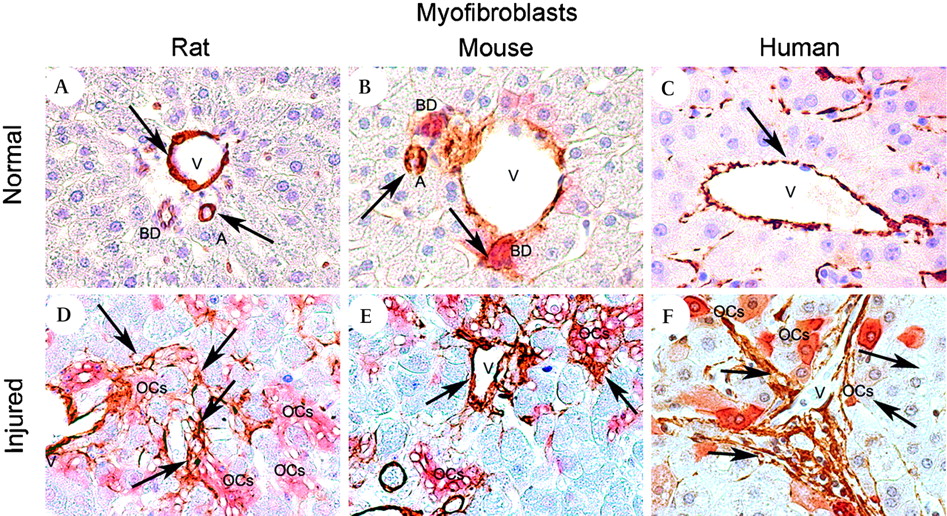
In normal liver tissue, αSMA+ myofibroblasts were seen only in very low number throughout the hepatic lobule, adjacent to bile ducts and in vessels walls (figure 2A–C). In both human and rodent injured liver an expanded population of αSMA+ myofibroblasts were seen periportally. Double staining confirmed an intimate association between myofibroblasts and the extending cords of OCs/HPCs (figure 2D–F), with myofibroblasts seen to line the columns of OCs/HPCs. Of note, the myofibroblasts formed a virtually continuous lining of the OC/HPC compartment, whereas macrophage association was more intermittent.

Myofibroblasts. Photomicrographs illustrating rodent and human liver tissue to visualise the partner cells of the hepatic stem cell niche. In control liver tissue (A–C) myofibroblast staining is localised mainly around the vessels (veins and arteries). (A) Normal rat liver, double staining for CK19 (red) and αSMA (brown); (B) normal mouse liver—double staining for panCK (red) and αSMA (brown); (C) control donor liver, αSMA (brown). In the injured liver (D–F) myofibroblasts form intimate contact with the bile ducts and the oval cells (OCs). (D) 2-Acetylaminofluorene and subsequent partial hepatectomy (2-AAF/PH) rat, day 9 after the PH—double staining for CK19 (red) and αSMA (brown); (E) hepatitis B surface antigen-tg-tg/retrorsine treated mouse—αSMA staining (brown); (F) hepatitis C virus-related cirrhotic liver—double staining for CK7 (red) and αSMA (brown). Magnification (A–E) ×40; (F) ×100.
As expected, vWF+ staining was demonstrated in the sinusoids and vessel walls of uninjured liver (figure 3A–C), In contrast, vWF+ vessels were seen periportally adjacent to OCs/HPCs in the injured tissue (figure 3D–F). A rich inflammatory infiltrate was seen, particularly in human chronic viral hepatitis where lymphoid follicles were surrounded by myofibroblasts and HPCs (figure 4A). Double staining showed that inflammatory infiltrate surrounding HPC was mainly composed of T (figure 4B) and B (figure 4C) lymphocytes.

Endothelial cells. Photomicrographs illustrating rodent and human liver tissue to visualise the partner cells of the hepatic stem cell niche. In control liver tissue (A–C) the endothelial cell-specific marker von Willebrand factor (vWF; brown) highlights veins and sinusoids. (A) Normal rat liver; (B) normal mouse liver; (C) control human donor liver. In the injured liver (D–F) endothelial cells (black arrows) appear in close contact with the bile ducts and the oval cells (OCs). (D) 2-Acetylaminofluorene and subsequent partial hepatectomy (2AAF/PH) rat, day 9 after the PH—vWF (brown); (E) hepatitis B surface antigen-tg/retrorsine treated mouse—vWF (brown); (F) biopsy specimen from hepatitis C virus cirrhotic liver—vWF (brown). Magnification (A–F) ×40.

Inflammatory infiltrate and hepatic progenitor cells (HPC) niche in humans with chronic hepatitis C. (A) Photomicrograph showing small inflammatory cells (black arrow) surrounded by HPC (red, CK7) and myofibroblast (brown, αSMA, yellow arrows) proliferation in the liver biopsy specimen of a patient affected by severe chronic hepatitis C. Specific lymphocyte marker showed that the inflammatory cells were mainly B lymphocytes (C, CD20, brown) while T lymphocytes (B, CD3, brown, black arrows) were less common (magnification ×40).
The niche in the HPC response during human liver disease: recurrent fibrosing cholestatic hepatitis and chronic viral hepatitis
We wished to study the cells associated with HPCs throughout the natural history of human liver disease, so we analysed liver tissue from a patient who had fibrosing cholestatic hepatitis due to hepatitis C recurrence after a liver transplant. Here, a pre-perfusion biopsy gave a baseline status (figure 5A). Over the next 6 months a marked ductular reaction developed owing to fibrosing cholestatic hepatitis (figure 5B). Sections serial to the HPCs were stained for macrophages (figure 5D) and myofibroblasts (figure 5E). These non-parenchymal cells were closely localised to the HPCs at both 1 (supplementary figure 2) and 6 months (figure 5C–E) after transplant. False colouring (figure 5F) of the three cell types from the 6-month serial sections with geographical alignment and overlay demonstrated the spatial co-localisation of the macrophages and myofibroblasts with the HPC. To confirm this observation, we performed double immunostaining as above in 10 further patients with chronic hepatitis C and 10 patients with chronic hepatitis B (figure 6). In both type of hepatitis, we confirmed the existence of a close contact between HPCs and myofibroblasts (figure 6A,D), HPCs and macrophages (figure 6B,E) and HPCs and laminin (figure 6C,F).
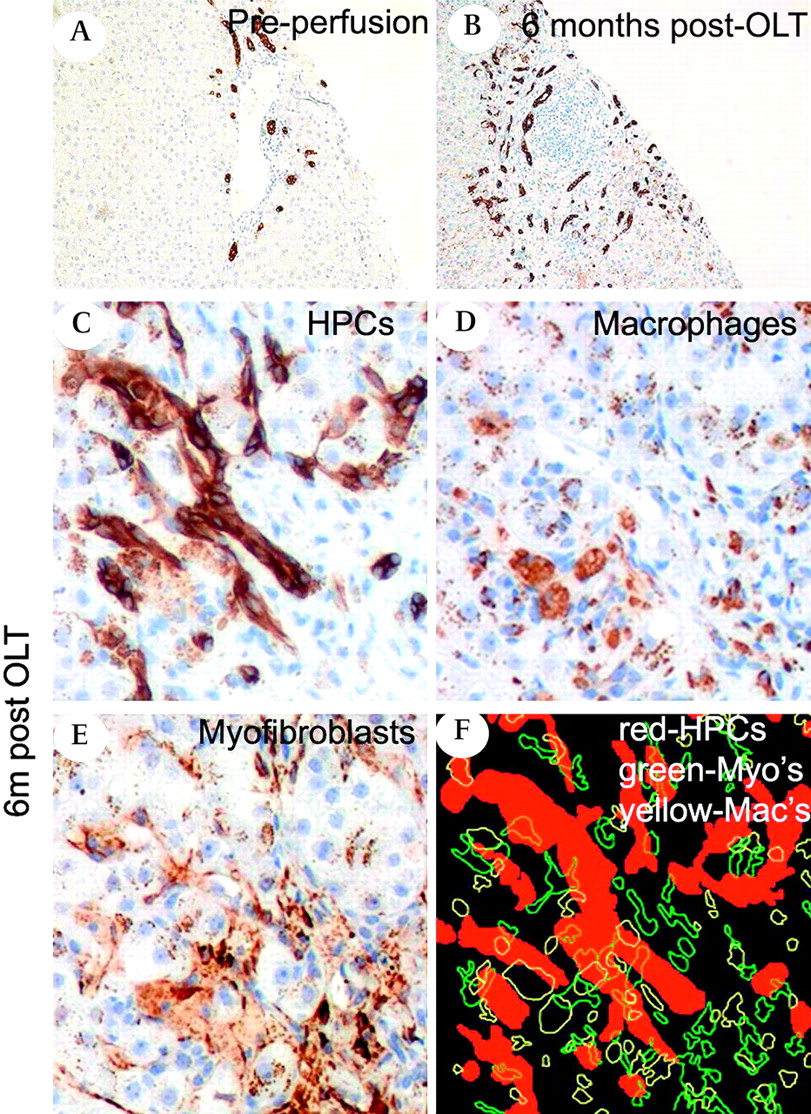
Liver progenitor cells rapidly expand in the human liver disease fibrosing cholestatic hepatitis. (A,B) Photomicrographs showing bile duct and hepatic progenitor cell (HPC) activation (CK7, brown) that has progressed rapidly in the liver of a patient who received a liver transplant for hepatitis C cirrhosis and developed fibrosing cholestatic hepatitis. (C–E) Serial sections from the 6-month biopsy reveal co-localisation of HPCs (CK7, brown), macrophages (CD68, brown) and myofibroblasts (αSMA). (F) In a composite image from the aligned sections, HPCs, macrophages and myofibroblasts are highlighted in red, yellow and green, respectively, and can be seen closely localised (magnification ×200). OLT, orthotopic liver transplantation.

A cellular niche consistently forms round progenitor cells in chronic viral hepatitis. Photomicrographs showing co-localisation of hepatic progenitor cells (HPC) with myofibroblasts (A,D), macrophages (B,E), and laminin (C,F) in chronic hepatitis B (HBV) and C (HCV) in humans. (A,D) CK7—red, αSMA—brown, black arrows indicating a myofibroblast lying HPC; (B,E) CK7—red; CD68—brown, black arrows indicating macrophages; (C,F) CK7—red, laminin—brown, black arrows indicating the laminin sheath (magnification ×100). These pictures are representative of the 10 patients with hepatitis C and of the 10 patients with hepatitis B whose liver biopsies were analysed.
Origin of the hepatic niche cells
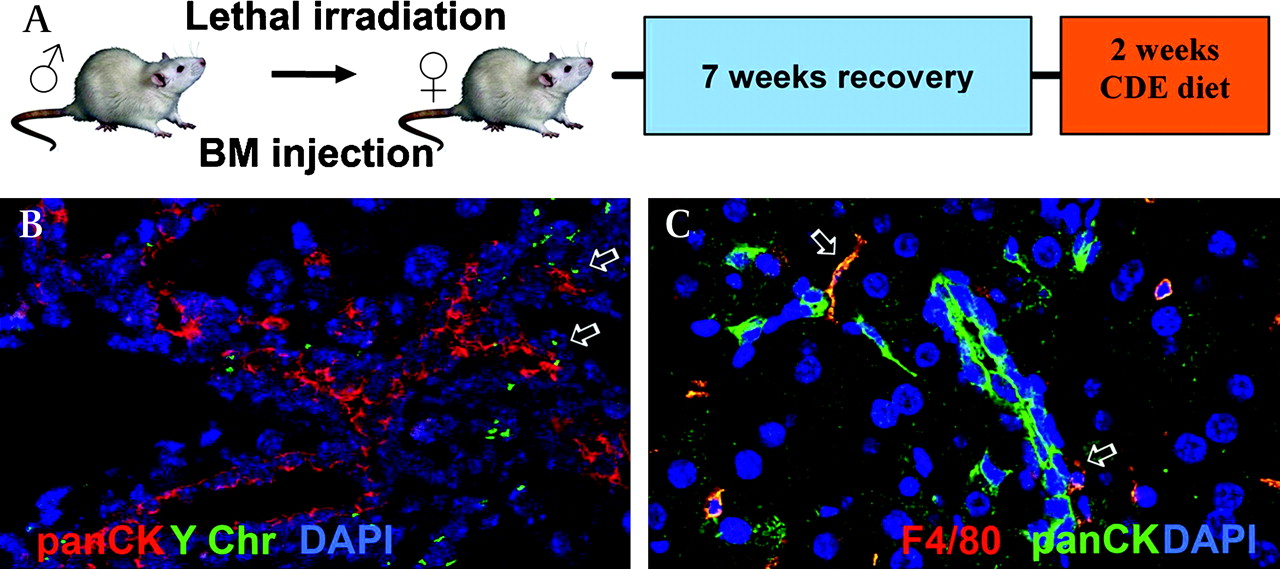
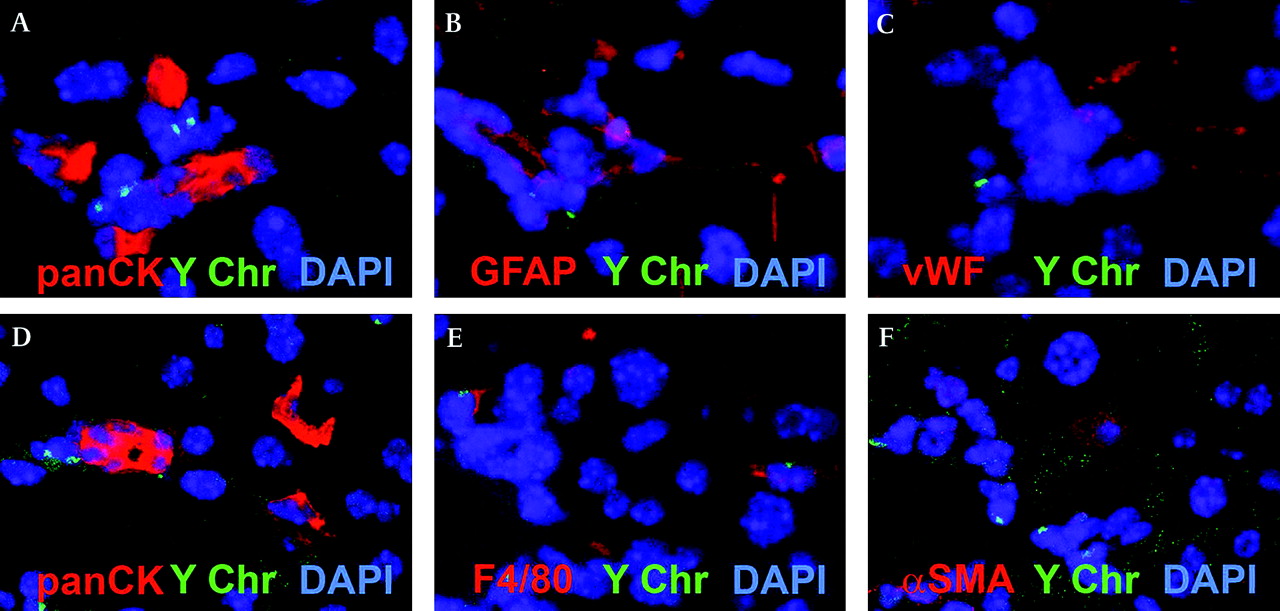
To investigate the BM origin of the OCs and the associated non-parenchymal niche cells, we performed sex-mismatched BM transplants in mice to allow BM cell tracking using in situ hybridisation for the Y chromosome. Previous studies suggesting that OCs do not have a BM origin have been criticised owing to the possibility that hepatotoxins (eg, retrorsine) used in the OC induction experimental protocol may inhibit the BM stem cell response. In this study, we therefore decided to use a dietary regimen, the choline-deficient ethionine-supplemented diet (CDE) alone to induce the OC response in mice (figure 7A). Our current results agree with our previous findings that OCs are not of BM origin20; however, immediately adjacent to the OCs we frequently observed BM-derived cells (figure 7B). Dual immunostaining for OC (panCK) and macrophage (F4/80) markers in this model demonstrated their immediate proximity (figure 7C). Confocal analysis of serial sections immunostained for NPC markers demonstrated that the F4/80+ macrophages are the predominant BM-derived cell population in the niche (figure 8E). In contrast to previous work looking at the origin of stellate cells and myofibroblasts in models of chronic liver fibrosis,17 although we did find Y-chromosome-positive stellate cells scattered throughout the parenchyma, we discovered that they did not co-localise with OCs in the present model of liver injury. It is possible that a longer period of injury would have produced a higher recruitment of myofibroblasts from the BM; however, this phenomenon appears to be injury dependent.

The origin of the oval cells in a mouse model of oval cell activation (choline-deficient ethionine-supplemented diet (CDE diet)). (A) Female mice were irradiated followed by injection of BM cells from a male donor. Seven weeks later the mice were treated with the ½ CDE diet for 2 weeks before tissue analysis. (B) Representative region of staining for oval cells (panCK, red) and in situ hybridisation for mouse Y chromosome (Y Chr, green). Arrows indicate Y chromosome-positive cells adjacent to panCK+ cells. (C) Double immunofluorescence for F4/80 (red) and panCK (green) in a mouse treated with the CDE diet for 2 weeks. Arrows indicate F4/80+ cells adjacent to panCK+ cells (magnification (B) ×10; (C) ×20).

The origin of the oval cell (OC)-associated niche cells in a mouse model of OC activation (choline-deficient ethionine-supplemented diet (CDE diet)). (A–C) and (D–F) Confocal analysis of serial sections of sex-mismatched mice treated with the CDE diet. Staining with panCK (A,D) demonstrated closely localised cells positive for GFAP (B), vWF (C), F4/80 (E) and αSMA (F). F4/80 but not GFAP, αSMA or panCK cells are Y chromosome positive (magnification ×400).
The matrix associated with the OC/HPC response is laminin rich
In rodent liver we found no relationship between collagen I and IV and the OCs (data not shown) but did find a striking relationship between laminin and the OCs in all rodent models of OC activation and human liver tissue studied. Laminin has previously been reported to be present in the basement membrane sheathing OCs. We found that laminin was present prominently around the portal vessels with weak staining in the hepatic sinusoids in the control liver (figure 9A–C). During liver injury, however, laminin was heavily deposited in a sheath which closely surrounds the OCs/HPCs (figure 9D–F), the OCs only extend as far as the laminin sheath (online supplementary figure 3A). Dual staining for laminin with OC and myofibroblast markers demonstrated that the laminin surrounded the OCs but co-localised with the myofibroblasts (supplementary figure 3D). However, in vitro cultures of isolated rat stellate cells and a rat OC line (LE6) confirmed that both cell types could produce laminin (supplementary figure 3E).
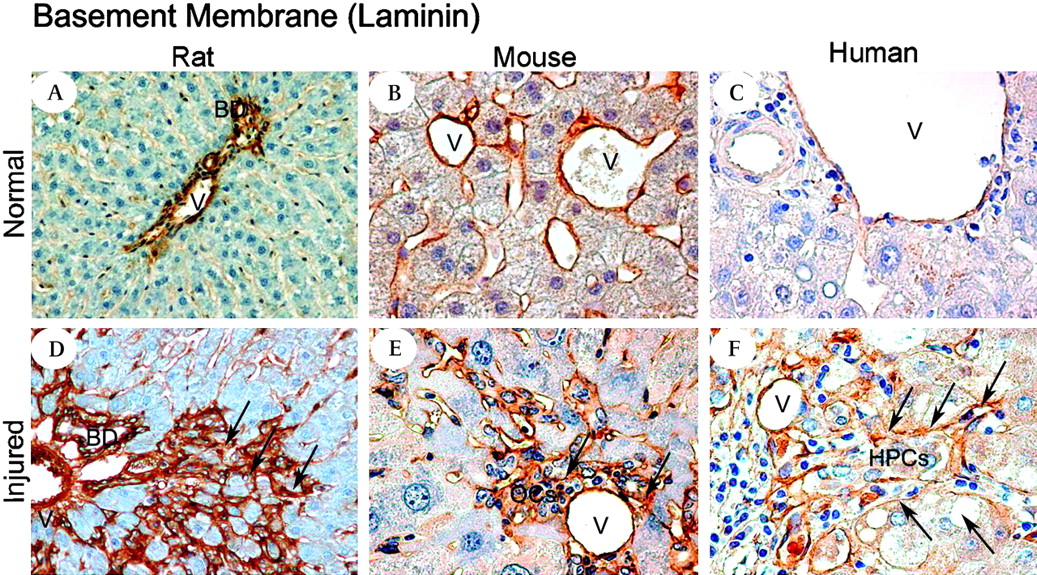
Laminin consistently surrounds liver progenitor cells in a variety of liver injury models and human disease. Photomicrographs illustrating laminin in the control liver (A, B, C) Normal rat, mouse and human pre-perfusion donor liver, respectively. A thin laminin layer (brown) is localised mainly around the bile ducts and the vessels. In the injured liver (D,E,F) laminin staining shows the appearance of a thicker membrane that spreads from the portal tract and builds a network-like structure intimately surrounding the oval cells /hepatic progenitor cells. (D) 2-Acetylaminofluorene and subsequent partial hepatectomy (2-AAF/PH) rat day 9 after the PH - laminin (brown, arrows); (E) hepatitis B surface antigen-tg/retrorsine treated mouse—laminin (brown, arrows); (F) hepatitis C virus-related cirrhotic liver—laminin (brown, arrows). (See also online supplementary figure 2.) Magnification (A,D) ×10; (B,C,E,F) ×40.
Laminin maintains a liver progenitor cell phenotype and aids biliary specification
We found that the extracellular matrix (ECM) strongly influenced the specification of OCs in vitro (figure 10). At baseline OCs were small rounded cells with high nuclear to cytoplasmic ratio which were panCK+. These cells were capable of proliferating for over 2 weeks in culture and increased in numbers (more than fourfold) between days 7 and 11. Isolated primary mouse OCs were cultured on plastic before switching to various ECMs. On plastic, initial cultures showed clusters of panCK+ HPCs (figure 10A). After replating on the matrices for 7 days no panCK+ cells persisted on collagen I and IV (figure 10B). Only cells on laminin maintained panCK+ cells (figure 10C), and these cells kept their original morphology. Only on the fibronectin matrix did we see cells of a hepatocyte morphology which were positive for the differentiated hepatocyte marker Cyp2D6 (figure 10D). Gene expression was strongly influenced by the matrices (figure 10E–H). Compared with the control starting cell population laminin profoundly upregulated progenitor (DLK1) and biliary (GGT, Aquaporin 1) genes while significantly inhibiting the early hepatocyte genes CEBP/a. In contrast, collagen I and IV and fibronectin inhibited or did not influence these progenitor and biliary genes but fibronectin strongly promoted CEPB/a gene expression. These results show that OCs have an ability to form hepatocytes and biliary epithelial cells that is influenced by the ECM.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The extracellular matrix determines oval cell (OC) cell behaviour. (A) Freshly isolated murine OCs showed high expression of panCK (red, nuclei blue). (B) After 1 weeks culture on collagen I or IV, no panCK+ cells persisted; (C) however, on laminin, panCK+ cells (panCK green, nuclei blue) were seen. No hepatocytes were differentiated from OCs on laminin. (D) Cells with a hepatocyte morphology (here binuclear and Cyp2D6+, red) were seen on the fibronectin matrix. (E,F,G) Laminin strongly promoted the gene expression of the biliary markers Aquaporin 1 (AQP1) (n=6, p<0.05) and GGT (n=6, p<0.005), and the liver progenitor marker DLK1 (n=6, p<0.05). (H) The early hepatocyte gene CEBP/a is found upregulated on fibronectin (n=6, p<0.0005); however, there is a significant reduction in CEBP/a gene expression when cells are cultured in the presence of laminin (n=6, p<0.0005). Values are relative to control cells grown on plastic just before replating onto the matrices (red dotted line).
Discussion
Liver regeneration usually occurs through hepatocyte division but in advanced or severe chronic liver disease, hepatocytes are unable to replicate efficiently and progenitor cells can regenerate the parenchyma.15 20 These OCs/HPCs have been well characterised but there is little data on their surrounding environment (or ‘niche’). In this study, we have systematically examined OC/HPC reactions in both rodent models of OC activation and in human liver disease. We elected to identify OCs/HPCs using cytokeratins as these are already widely accepted HPCs markers and as we have demonstrated, in accordance with previous studies, are analogous between species and models.10 27 Furthermore here we demonstrate that these cells represent a progenitor cell population which, while potentially heterogeneous, contains cells with the ability to form both hepatic epithelial lineages.
We found that the OCs/HPCs are closely accompanied by a cellular niche composed of myofibroblasts, macrophages and endothelial cells throughout the progenitor cell response, which remains consistent throughout a range of diverse rodent models of OC activation and also in human liver disease. We sought to identify the origin of the niche cells using genetic tracking techniques in a dietary model of OC activation. This confirmed that in a dietary model of OC activation, without retrorsine administration, OCs/HPCs are definitely of intrahepatic origin; they are, however, intimately surrounded by macrophages of BM origin. This provides a potential link between the BM and the intrahepatic stem/progenitor cells during injury, whereby the BM does not directly contribute to these cells but may influence their behaviour. In contrast with our previously published study,17 only a few scattered myofibroblasts were found to be of BM origin in the CDE diet model of liver injury and these cells did not localise with the OCs. It may be that longer injury would increase the recruitment of cells from the BM or, alternatively, the mode of liver injury may influence the magnitude of recruitment.
The exact role of these progenitor-associated macrophages and myofibroblasts is unknown; however, these NPCs may act as a link between tissue injury and progenitor cell activation. Indeed, hepatic macrophages and myofibroblasts can express a variety of signals critical for controlling liver development and hepatic stem/progenitor cell behaviour.27 Macrophages can also remodel the ECM through the production of metalloproteinases,30 a process that may be necessary for the extension of the OC/HPC response.
In addition to signals passing from the niche cells to the progenitor cells, there may also be signalling from the progenitor cells to the niche cells. We hypothesise that the profound progenitor cell response seen in severe chronic liver disease, such as fibrosing cholestatic hepatitis after transplantation may account for the local activation of myofibroblasts and macrophages and thus may help to explain the extremely rapid fibrosis in this condition; however, this hypothesis will require further evaluation.
Little is known about the influence of the ECM upon OC function but the uniform presence of laminin around OCs suggests an important role. We hypothesised that laminin was influencing the progenitors and allowing maintenance of the undifferentiated phenotype. Indeed our previous studies have found that laminin matrix promotes the proliferation and expansion of OCs in vitro.31 We sought to investigate this in a controlled in vitro system. Using isolated primary murine OCs, we found that laminin, unlike the other matrices tested, permitted the culture of OCs in an undifferentiated phenotype. Laminin also promoted gene expression of biliary/OC genes and significantly inhibited expression of the early hepatocyte gene CEBP/a. These findings are consistent with a key role for laminin in controlling liver progenitor cell fate.
In summary, we have shown using independent rodent models of severe liver injury and human disease that a niche composed of myofibroblasts, macrophages and laminin always surrounds the progenitor cells. The OCs are definitively of intrahepatic origin and do not derive from the BM, but macrophages of BM origin intimately associate with the OCs. The signalling pathways linking OCs/HPCs and the cells in the niche are likely to be bidirectional. Simultaneous alteration in matrix composition, activation of resident non-parenchymal cells and activation of intrahepatic progenitor cells are features of liver regeneration. Future work will therefore focus on these matrix–progenitor cell and niche–progenitor cell interactions in order to develop therapeutic interventions in chronic liver diseases.
Significance of this study
What is already known about this subject?
Hepatic progenitor cells are activated in chronic liver injury and contribute to liver regeneration.
Hepatic progenitor cells are bipotential and can give rise to hepatocytic and biliary epithelial cells.
What are the new findings?
A stereotypical progenitor cell niche, composed principally of macrophages, myofibroblasts and laminin matrix, develops in many types of chronic liver injury.
This niche can develop rapidly in fibrosing cholestatic hepatitis and may contribute to the rapid development of fibrosis seen in this condition.
The laminin matrix enables hepatic progenitor cells to remain in an undifferentiated phenotype.
How might it impact on clinical practice in the foreseeable future?
Novel targets to control progenitor cell behaviour may be identified.
The targeting of progenitor cells using drugs or other techniques may influence both liver regeneration and fibrosis.
Acknowledgments
SJF is supported by the Medical Research Council, Wellcome Trust and Jules Thorn Trust. SL is supported by European Association for the Study of the Liver (EASL) Sheila Sherlock Post-Doc Fellowship and by “Ordine dei Medici Chirurghi ed Odontoiatri di Bologna”. TGB is supported by a Wellcome Trust Clinical Training Fellowship. The authors wish to thank Professor Roland Wolfe for the kind provision of the Cyp2D6 antibody and Professor Nelson Fausto for kindly supplying the LE/6 cell lines used in this study.
References
Footnotes
Funding Medical Research Council. Other Funders: Wellcome Trust.
Competing interests None to declare.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Digest