Article Text

Abstract
Background: The trisubstituted methylxanthine derivative pentoxifylline inhibits hepatic stellate cell proliferation and collagen synthesis in vitro. The antifibrotic effect of pentoxifylline in a suitable in vivo model of chronic liver fibrogenesis remains to be tested.
Methods: Groups of adult rats (n=20–23) received oral pentoxifylline at a dose of 8 mg/kg/day from week 1 to week 6, and 16 mg/kg/day from week 1 to week 6 or week 4 to week 6 after complete bile duct occlusion. Animals who underwent sham operation that received 16 mg/kg/day pentoxifylline and untreated rats with bile duct occlusion alone served as controls. After six weeks, animals were sacrificed and parameters of fibrogenesis determined.
Results: Bile duct occlusion caused portal cirrhosis with a 10-fold increased hepatic collagen content in the absence of inflammation or necrosis. This was accompanied by an 11-fold elevated serum aminoterminal procollagen III peptide (PIIINP). The drug induced a dramatic eightfold downregulation of procollagen I mRNA, and suppression of the fibrogenic factors transforming growth factor β1 and connective tissue growth factor by 60–70%. However, profibrogenic tissue inhibitor of metalloproteinase 1 (TIMP-1) mRNA was increased twofold, resulting in only a moderate decrease in liver collagen, fibrosis score, and PIIINP.
Conclusions: We conclude that targeting pentoxifylline to the fibrogenic cells, thereby avoiding upregulation of TIMP-1, could become a potent antifibrogenic tool in chronic liver disease.
- antifibrotics
- pentoxifylline
- cirrhosis
- connective tissue growth factor
- ALT, alanine aminotransferase
- ALP, alkaline phosphatase
- AST, aspartate aminotransferase
- BDO, bile duct occlusion
- cAMP, cyclic adenosine monophosphate
- CTGF, connective tissue growth factor
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- γGT, gamma glutamyl transpeptidase
- HYP, hydroxyproline
- PCR, polymerase chain reaction
- PDE, phosphodiesterase
- PTX, pentoxifylline
- PIIINP, (serum) aminoterminal propeptide of procollagen type III
- TGF-β, transforming growth factor β
- TIMP-1, tissue inhibitor of metalloproteinase 1
Statistics from Altmetric.com
- ALT, alanine aminotransferase
- ALP, alkaline phosphatase
- AST, aspartate aminotransferase
- BDO, bile duct occlusion
- cAMP, cyclic adenosine monophosphate
- CTGF, connective tissue growth factor
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- γGT, gamma glutamyl transpeptidase
- HYP, hydroxyproline
- PCR, polymerase chain reaction
- PDE, phosphodiesterase
- PTX, pentoxifylline
- PIIINP, (serum) aminoterminal propeptide of procollagen type III
- TGF-β, transforming growth factor β
- TIMP-1, tissue inhibitor of metalloproteinase 1
Curative or preventive treatment is only available for a minority of patients with chronic liver disease. Currently, there is no established therapy to halt the progression of liver fibrosis or to induce regression of cirrhosis. In order to prove an in vivo antifibrotic effect of drugs that can suppress collagen synthesis in vitro, animal models that resemble human chronic liver diseases have to be used.1 Complete occlusion of the extra and intrahepatic biliary system in rats generates a progressive portal fibrosis and, ultimately, cirrhosis, that is essentially devoid of hepatocyte necrosis or major inflammation.2,3 This model is suitable for detecting drugs that are truly antifibrotic in vivo, at least in biliary fibrosis, the effect of which is not obscured by their activities as radical scavengers or antiphlogistics, both properties which have been shown to be ineffective in preventing the progression of fibrosis in humans.4–6
Pentoxifylline (PTX) is a trimethylated xanthine derivative which is widely used in disorders of vascular perfusion due to its favourable effects on erythrocyte deformability, erythrocyte oxygen delivery, and its properties as a peripheral vasodilator.7,8 PTX also improves liver perfusion in humans.9–11 As with other methylxanthines, PTX is an inhibitor of phosphodiesterases, resulting in an elevated intracellular pool of the second messenger cyclic adenosine monophosphate (cAMP).12 In vitro studies with fibroblasts have shown that PTX potently reduces cell proliferation, stimulates interstitial collagenase activity, and suppresses the synthesis, secretion, and deposition of fibrillar collagens type I and III, proteoglycans, and fibronectin.13–17 More recently, an antifibrogenic effect of PTX on activated hepatic stellate or myofibroblast-like cells, which are responsible for the excessive extracellular matrix production in liver fibrosis,18–20 has been demonstrated.21–24 These studies concluded that PTX might exert its in vitro antifibrogenic effect on hepatic stellate cells primarily by inhibiting procollagen I and III mRNA expression and by downregulating the stellate cell mitogen platelet derived growth factor, events which were correlated with an increase in cellular levels of cAMP.
The activities of PTX, affecting cell membranes, cytokine and growth factor signalling, as well as mesenchymal cell proliferation and collagen synthesis, coupled with its well explored safety profile, make testing of its antifibrotic properties in the more complex context of a suitable in vivo model of progressive hepatic fibrosis mandatory. In a previous study, using a model of porcine liver fibrosis induced by feeding yellow phosphorus, PTX reduced the increased liver collagen content only moderately, from 144% to 113% of controls.25 As the number of animals per group was low (n=3), the model is pathophysiologically unrelated to human liver fibrosis, and as expression of profibrogenic genes was not investigated, interpretation of the data is difficult.
Using the model of chronic rat secondary biliary fibrosis, we demonstrated that doses of PTX comparable with those administered for human vascular disorders caused an unexpected eightfold downregulation of hepatic procollagen type I mRNA, and significant suppression of hepatic expression of two central fibrogenic cytokines, transforming growth factor β1 (TGF-β1)19,20 and connective tissue growth factor (CTGF).26 However, a concomitant increase in tissue inhibitor of metalloproteinase-1 (TIMP-1) mRNA reduced the antifibrotic effect of PTX. Derivatives of PTX targeted to the fibrogenic cells of the liver or that lack TIMP-1 upregulating activity could become potent antifibrotic agents in chronic liver diseases.
METHODS
Rat model of secondary biliary cirrhosis
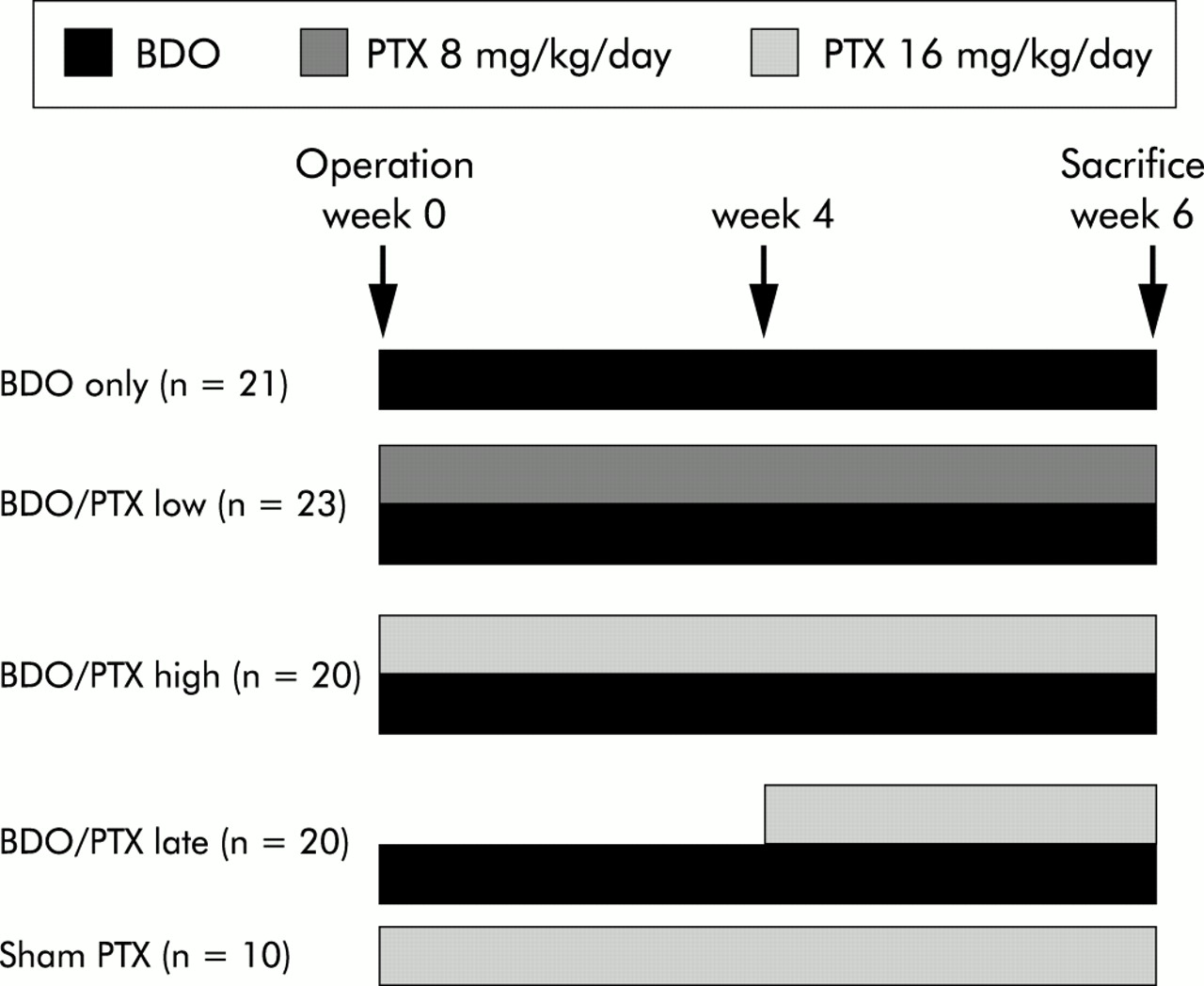
Adult female Wistar rats (Schoenwalde, Germany) weighing 206 (±19) g were maintained in 12 hour light/dark cycles at 23 (±2)°C, with 60 (±10)% humidity. Complete bile duct occlusion (BDO) was performed as reported previously.2,3 Briefly, after midline abdominal incision and proper isolation, the common bile duct was occluded by injection of sodium amidotriazoate (Ethibloc 0.2 ml/kg body weight; Ethicon, Germany) in a retrograde direction using a Teflon catheter (Abbocath-T 26 G; Abbott, Chicago, Illinois, USA), followed by double ligation and scission in-between. Sham operation was a midline abdominal incision, isolation of the common bile duct, and wound closure. PTX (Trental; Aventis, Germany) was added to the drinking water and did not alter fluid consumption by the animals. The following therapeutic groups were formed: (1) BDO and PTX at 16 mg/kg/day for six weeks (n=20); (2) BDO and 8 mg/kg/day PTX for six weeks (n=23); and (3) BDO and PTX at 16 mg/kg/day from week 4 to week 6 (n=20). Sham operated animals that received PTX at 16 mg/kg/day (n=10) and bile duct occluded animals without treatment (n=21) served as controls (fig 1). After six weeks, animals were sacrificed and aliquots of right and left liver lobes snap frozen in liquid nitrogen. All animal experimentation was carried out in accordance with our institutional and governmental regulations on the use of experimental animals. After six weeks, rats were sacrificed under anaesthesia by puncture of the right ventricle and exsanguination. Heart, liver, spleen, and kidneys were weighed, and 1–2 g pieces of the left and right liver lobes fixed in 4% formalin for histology and hydroxyproline (HYP) determinations. Early mortality (within one hour to three days) of rats with BDO due to local infection was 9%. As significant fibrosis becomes evident only after two weeks of BDO, these animals were not considered for statistical analysis.

Study design. BDO, bile duct occlusion; BDO/PTX low, BDO/PTX high, BDO/PTX late, rats with BDO treated with pentoxifylline (PTX) at 8 mg/kg/day from week 1 to 6, at 16 mg/kg/day from week 1 to week 6, or with PTX at 16 mg/kg/day from week 4 to week 6, respectively. Rats who underwent sham operation and daily treatment with PTX at 16 mg/kg/day from week 1 to week 6 (sham PTX) served as controls.
Histological scoring
For each liver, 1 μm paraffin sections of the right and left lobe were stained with haematoxylin/eosin, trichrome (Masson-Goldner), and silver impregnation (Gomori), and scores (see below) of both lobes averaged. Inflammation and necrosis were graded according to the histological activity index of Knodell and colleagues.27 Staging of fibrosis followed the slightly modified method of Ruwart and colleagues28 which, contrary to conventional scores, is particularly suited for detecting slight changes in portal fibrosis: normal liver (0 points); increased collagen with some periportal stellate bile duct proliferations (1 point); increased collagen with incomplete septa that do not form porto-portal or porto-central connections (2 points); strongly increased collagen, more incomplete than complete septa (2.25 points); an equal number of incomplete and complete septa (2.5 points); more complete than incomplete septa (2.75 points); only complete septa (3); complete septa and less than 25% diffuse parenchymal fibrosis (3.25 points); complete septa and 25–50% diffuse parenchymal fibrosis (3.5 points); complete septa and 50–75% diffuse parenchymal fibrosis (3.75 points); and complete septa and more than 75% diffuse parenchymal fibrosis (4 points). Point scores from each animal were derived from the means of the right and left liver lobes.
Determination of hepatic hydroxyproline and serum aminoterminal procollagen III peptide
Hepatic HYP content was determined in duplicate from approximately 0.2 g hydrolysed tissue from the right and left liver lobes using the method of Jamall and colleagues29 with minor modifications.2,3 Liver collagen content was calculated from mean HYP concentration of both lobes (both lobes differed by less than 8%). Serum aminoterminal procollagen III peptide (PIIINP) was measured by a radioimmunoassay based on rat PIIINP, a monospecific rabbit antiserum to rat PIIINP and a goat antiserum to rabbit IgG, as detailed previously.2,3,30
cDNA probes and multiprobe RNase protection assay
A 1.3 kb PstI/HindIII fragment of plasmid α1R1 containing cDNA encoding rat procollagen α1(I), kindly provided by Dr D Rowe (Department of Pediatrics, University of Connecticut Health Center, Farmington, Connecticut 06032, USA),31 was subcloned into pGEM1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)32 and TGF-β133 plasmids were gifts from Dr XL Tian (Max Delbrueck Centre for Molecular Medicine, Berlin-Buch, Germany). For generation of all other probes, rat liver total RNA was reverse transcribed with SuperscriptII Reverse Transcriptase (Gibco Life Technologies, Freiburg, Germany) and oligo-dT primer according to the manufacturer's instructions. The cDNA for rat TIMP-1 was generated by polymerase chain reaction (PCR) using a mixture of Taq and Pfu DNA polymerases (Gibco and Stratagene, Amsterdam, the Netherlands) and primers binding to positions 109–133 and 415–439 of the published sequence.34 Similarly, rat CTGF cDNA was amplified with primers caa ccg caa gat tgg agt gt and ctc cag tct gca gaa ggt att g according to positions 398–417 and 806–827 of the mouse CTGF sequence (fisp-12, GenBank accession No M70642).35,36 PCR products were cloned into the EcoRV site of pZErO-1 and sequenced on both strands. The rat specific sequence of the CTGF amplicon has been submitted to GenBank/EMBL Data Libraries with accession No AJ236872. The rat protein displays 95% homology to the mouse, human, and porcine protein.
Liver total RNA was extracted using the acid guanidinium thiocyanate-phenol-chloroform method,37 and its integrity documented by visualisation of 18S and 28S ribosomal bands after electrophoresis. Radiolabelled RNA probes were produced by in vitro transcription with T7 polymerase (Ambion, Austin, Texas, USA) using [α-32P] UTP (800 Ci/mmol, 10 mCi/ml; NEN, Boston, Massachusetts, USA). Specific activity of all radiolabelled transcripts was usually about 10×105 cpm/μl. RNA was incubated with 2.0–4.0×105 cpm of 32P labelled RNA probes, denatured at 90°C, and hybridised overnight at a temperature optimised by preliminary experiments (range 43–46°C). After hybridisation, RNAse T1 (Ambion) was added to digest unbound label and unprotected mRNA. The protected RNA-RNA hybrids were denatured and separated by electrophoresis through a 5% polyacrylamide/urea sequencing gel. Gels were exposed to x ray films for 16–24 hours, and autoradiographs analysed with the public domain NIH Image program. Signals for procollagen α1(I) and TIMP-1 mRNA were normalised to the GAPDH mRNA signal and expressed as relative abundance (arbitrary units). Expected RNAse protected transcript sizes were as follows: procollagen α1(I) 230 bp (from BamHI to RsaI site, M11432, M12198, and M12199); TIMP-1 264 bp (from position 176 to 439 bp, L29512); CTGF approximately 430 bp (the rat CTGF sequence homologous to positions 398–827 of mouse CTGF); TGF-β1 253 bp (from position 1197 to 1449 in X52498); and GAPDH 102 bp (from position 335 to 436 bp, M17701).
Statistics
Data are presented as mean (SD) and median (25th/75th percentiles). Statistical analysis was performed using the Mann-Whitney rank sum test and ANOVA. Differences in relative abundance of mRNAs were analysed using the Kruskal-Wallis test.
RESULTS
A total of 84 of 93 rats survived beyond day 4 after BDO (early mortality 9%, with no mortality thereafter). Groups of 20–23 surviving rats received either no PTX (untreated BDO controls), PTX at a dose of 8 mg/kg/day (low), or 16 mg/kg/day (high) for six weeks, or PTX (high) from week 4 to week 6 of BDO. Ten sham operated rats given PTX (high) served as normal controls (fig 1). All rats with BDO became icteric after a few days and displayed severe secondary biliary fibrosis at sacrifice. Body weights increased in all groups over the six weeks but were significantly lower only in rats with BDO that received PTX (high) (table 1). Liver and spleen weights were elevated 2–3-fold in all rats with BDO compared with non-fibrotic controls. However, animals with BDO that received 16 mg PTX exhibited significantly lower liver and spleen weights than the other groups with BDO (p<0.01) (table 1). Similarly, heart and kidney weights were increased in all fibrotic animals (p<0.01), and again lower in the group that received PTX (high) compared with untreated rats with BDO alone (p<0.05, not shown).
Clinical, chemical, and fibrosis parameters in treated and untreated groups of rats
Total hepatic collagen content was determined as HYP. BDO for six weeks increased total liver collagen 10-fold (fig 2A). PTX (high) over six weeks reduced liver collagen by 24% (p<0.01). But there was only a trend towards collagen reduction with PTX (low) (p=0.06), and no antifibrotic effect in the group that received PTX (high) from week 4 to week 6—that is, when treatment was started at an already fourfold increased total liver collagen.2,3 The relative proportion of collagen (in mg/g liver), which increased threefold after six weeks of BDO, showed a reduction only in the group of rats that received PTX (high) over six weeks (p<0.05) (fig 2B). The quantitative collagen data were confirmed by histological staging using a refined scoring system. Here, the median of the untreated group with BDO reached 3.5 points compared with 2.75 in the group with PTX (high) over six weeks (p<0.05), and 3.25 points in the group with PTX (high) from week 4 to week 6 (NS) (fig 2C). There was no evidence of inflammation and necrosis in any group of fibrotic rats (score of 0). To assess the effect of PTX on hepatic stellate cell/myofibroblast activation, sections were stained for desmin. When we counted the number of desmin positive cells in three high power fields both of (fibrotic) portal tracts and (non-fibrotic) centrilobular areas of each liver, we did not detect significant differences between groups with BDO (not shown).

Effect of pentoxifylline (PTX) on parameters of liver fibrosis of normal and bile duct occluded (BDO) rats. (A) Total liver collagen expressed as total hydroxyproline. (B) Relative content of collagen, as hydroxyproline per gram of wet liver weight. (C) Modified histo score. (D) Serum aminoterminal procollagen type III peptide (PIIINP). Sham 16 w1–6, sham operation and PTX at 16 mg/kg/day for six weeks; BDO w1–6, bile duct occlusion for six weeks; BDO 16 w1–6, bile duct occlusion and PTX at 16 mg/kg/day for six weeks; BDO 8 w1–6, bile duct occlusion and PTX at 8 mg/kg/day for six weeks; BDO 16 w4–6, bile duct occlusion for six weeks and PTX at 16 mg/kg/day from week 4 to week 6. Significant differences were calculated using the Mann-Whitney rank sum test, and data are presented as box plots, with medians and boxes representing the 25th and 75th percentiles. *p<0.05, **p<0.01 versus the untreated group (BDO alone).
At sacrifice, serum levels of PIIINP, a surrogate marker of liver fibrogenesis, were increased 11-fold after BDO compared with the sham operated controls. PIIINP was lowered significantly in both groups that received PTX over six weeks (p<0.05) but unchanged in the group treated from week 4 to week 6 (fig 2D).
Hepatic procollagen α1(I), TIMP-1, TGF-β1, CTGF, and GAPDH steady state mRNA levels were determined by multiprobe ribonuclease protection assays. The protected bands showed the expected sizes and there were no equivalent bands in the negative controls. After six weeks of BDO, hepatic procollagen α1(I), TIMP-1 and TGF-β1 mRNA, normalised to GAPDH mRNA, were highly increased relative to the sham operated controls (all p<0.001) (fig 3). PTX downregulated the 20-fold increased procollagen α1(I) mRNA by a factor of eight (p<0.001) whereas it upregulated TIMP-1 mRNA twofold (p<0.05). TGF-β1 and CTGF transcript levels were decreased by approximately 60–70% (fig 3).

Modulation of hepatic procollagen α1(I) (Pro α1 (I)), tissue inhibitor of metalloproteinase 1 (TIMP-1), transforming growth factor β (TGF-β1), and connective tissue growth factor (CTGF) mRNA expression by pentoxifylline (PTX) treatment. RNA from livers of five rats in each experimental group was extracted and analysed for pro α1 (I), TIMP1, TGF-β1, CTGF and GAPDH mRNA content by multiprobe RNAse protection assay, followed by densitometry of protected bands. (A) Representative autoradiographs. (B) Results of the densitometrical analysis normalised to GAPDH. Sham/PTX, sham operation and PTX (high) for six weeks; BDO, bile duct occlusion alone for six weeks; BDO/PTX, bile duct occlusion and PTX (high) for six weeks. Data are mean (SD). *p<0.05, **p< 0.001 versus untreated group (BDO alone).
Alkaline phosphatase (ALP), gamma glutamyl transpeptidase (γGT), and bilirubin were increased 3.5, 9, and 27-fold, respectively, in the BDO compared with the sham operated animals. These cholestasis parameters were not affected by treatment with PTX (table 1). Similarly, aspartate aminotransferase (AST) and alanine aminotransferase (ALT), reflecting inflammation and necrosis, did not differ among groups except for a moderate elevation in AST in rats that received both doses of PTX for six weeks (p<0.05).
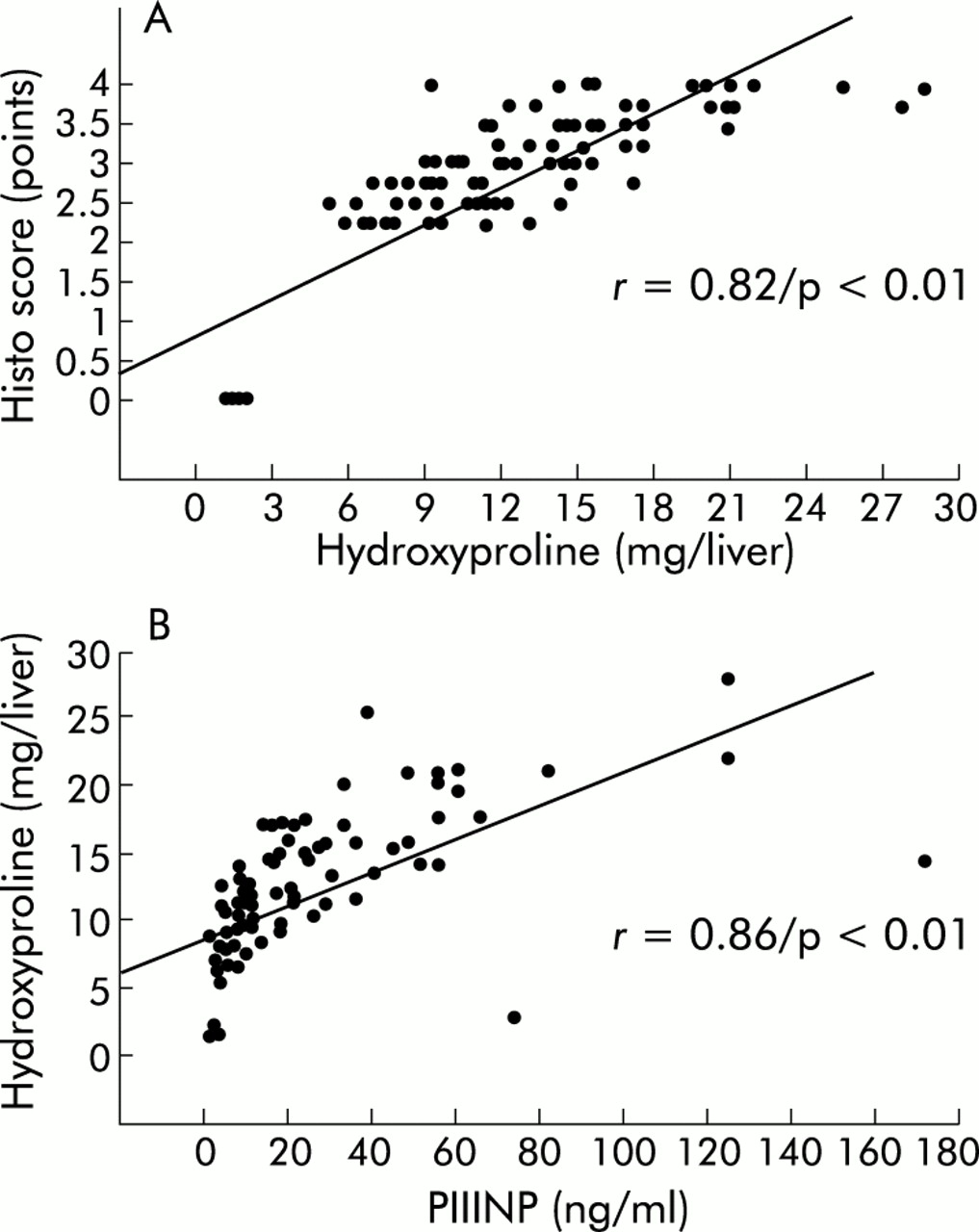
To investigate if one of the serum parameters reflected the decrease in hepatic collagen accumulation in animals treated with PTX, correlations were calculated using the Spearman rank order test (table 2). Liver hydroxyproline correlated well with histological stage (p<0.01) (fig 4A), and PIIINP showed a good correlation with histological staging (p<0.01) and total liver collagen (p<0.01) (fig 4B). Furthermore, the parameters of liver fibrosis (HYP content, histo score) and fibrogenesis (PIIINP) correlated well with liver and spleen weights (table 2) but not with serological markers of inflammation and necrosis (AST, ALT), kidney function (serum creatinine), or cholestasis (ALP, bilirubin, γGT).
Correlations between the investigated parameters. Coefficients were calculated using the Spearman rank order correlation, and highly significant correlations (p<0.01) are printed in bold

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Correlations between total liver collagen, aminoterminal propeptide of procollagen type III (PIIINP), and the histo score. Correlations were calculated using the Spearman rank order correlation, as described in the methods.
DISCUSSION
We previously showed that experimental liver fibrosis due to complete obstruction of the biliary tract is a suitable model for testing drugs that inhibit hepatic fibrogenesis in vivo.3,4 This model is characterised by a progressive portal fibrosis accompanying the proliferating bile ducts. As inflammation and necrosis are virtually absent, a significant reduction in liver collagen in this model should allow better selection of true antifibrotic and not merely anti-inflammatory or radical scavenging agents that could be useful in human liver fibrosis.
Several studies showed that the methylxanthine derivative PTX, an inhibitor of phosphodiesterases, can inhibit mesenchymal cell proliferation and collagen synthesis in vitro.14–17 Most of these investigations had been performed with dermal fibroblasts but recently several groups confirmed these effects of PTX in rat21 and human hepatic stellate and myofibroblast cell cultures.22–24
Previous in vivo studies used porcine liver fibrosis subsequent to feeding of yellow phosphorus25 which bears little similarity to human disease, or rat carbon tetrachloride induced fibrosis, a model that involves free radicals and severe necroinflammation of the pericentral zone.38 Importantly, the numbers of animals in these studies were low. A recent report investigated the effect of a single intraperitoneal injection of PTX (50 mg/kg body weight) in rats 24 hours prior to bile duct ligation for only three days.39 The authors demonstrated an antiproliferative effect of PTX on hepatic myofibroblasts in vivo but did not investigate fibrosis. A single study suggested no significant antifibrotic effect of PTX (50 mg/kg/day for four weeks) in rats with bile duct ligation.40 However, lower numbers of animals were used, fibrosis and cholestasis were less advanced, and apart from morphometry, no biochemical collagen determinations or investigations of matrix metabolism were carried out. Therefore, we studied the antifibrotic efficacy of PTX and parameters of matrix turnover in our model of chronic BDO.
We showed that PTX, when given orally at a dose of 16 mg/kg/day, reduced total hepatic collagen accumulation moderately but significantly whereas the effect of half the dose did not reach statistical significance. In contrast with its moderate antifibrotic effect, PTX potently downregulated hepatic steady state levels for procollagen α1(I) mRNA, which encodes the major collagen deposited during fibrogenesis, from 20-fold to 2.5-fold above baseline in bile duct obstructed rats. This eightfold reduction in procollagen α1(I) mRNA is the most dramatic effect of a drug on procollagen I expression in this progressive model of fibrosis and, to our knowledge, unmatched in any other in vivo model of hepatic fibrosis. It was shown before that apart from being an antiproliferative drug for mesenchymal cells, PTX also potently suppresses interstitial procollagen expression in vitro.17,41 In rat biliary fibrosis, only silymarin, a mixture of well defined flavonoids, and LU135252, a specific endothelin A receptor antagonist, inhibited collagen accumulation and procollagen α I mRNA expression by up to 60%.3,4 However, in contrast with these two drugs, PTX upregulated expression of TIMP-1, the most important endogenous inhibitor of most matrix metalloproteinases, more than twofold in rats with BDO, whereas silymarin and LU135252 suppressed TIMP-1 three and twofold, respectively. These findings strongly suggest that in vivo, upregulation of TIMP-1 expression by PTX counteracts its powerful suppressive effect on procollagen I expression, resulting in the observed modest net reduction in fibrosis. While PTX downregulates TIMP-1 in fibroblasts and culture activated hepatic stellate cells,24 it could upregulate TIMP-1 mRNA expression in hepatocytes, bile duct epithelia, and Kupffer cells which do not express procollagen I but significant amounts of TIMP-1 mRNA during fibrogenesis.42,43 Thus our findings caution against extrapolation of in vitro data using activated stellate cells or myofibroblasts alone for the far more complex in vivo situation of fibrogenesis.
The effect of PTX on procollagen I mRNA expression is even more remarkable when we consider that the higher dose of the drug did not exceed the upper dose that is usually prescribed for humans to improve the macro and microcirculation.9 The published plasma levels at this oral dose range from 2 to 5 μg/ml44 and are thus below concentrations that are required to suppress collagen synthesis of activated hepatic stellate cells and myofibroblasts in vitro, reaching 27% (at 100 μg/ml) or 67% (at 500 μg/ml).23 In other in vivo experimental settings such as thrombin induced platelet activation,44 this was explained by an as yet undefined in vivo accumulation or activation of the drug. At the applied dosage, PTX did not cause hepatic inflammation or necrosis while AST was slightly elevated in the groups that received PTX over six weeks (table 1). Similarly, PTX did not change the laboratory parameters of cholestasis, suggesting that its antifibrotic properties are unrelated to an effect on hepatobiliary function but rather due to a direct impact on the effector cells of hepatic fibrogenesis—that is, activated stellate cells and (portal tract derived) myofibroblasts. This is supported by Desmouliere and colleagues39 who found significantly reduced myofibroblast proliferation (PCNA index) in their short term model of BDO. In line with our data, they also found that PTX did not reduce the number of desmin positive myofibroblasts. We are confident that by using desmin instead of α-actin as a marker of activated HSC in rat liver fibrosis, the majority of activated stellate cells/myofibroblasts will be labelled. Thus according to an in depth study by Tuchweber and colleagues,45 desmin and α-actin positivity go almost hand in hand in later stages of biliary fibrosis, the model that we used in this study.
PTX significantly lowered hepatic mRNA expression of TGF-β1 and CTGF in rats with biliary fibrosis, two key mediators of fibrogenesis that act in concert19,20,36,46 to enhance fibroblast collagen and TIMP-1 expression. This suggests that at least part of the antifibrogenic effect of PTX may be due to downregulated expression of these profibrogenic cytokines.
Suppression of stellate cells and myofibroblasts by PTX appears to involve at least three different mechanisms that act in part downstream of an increase in intracellular cAMP13,22,47: (1) inhibition of procollagen I and III expression at a pretranslational level by downregulation of nuclear factor 1, a positive transcriptional regulator of interstitial procollagen genes, which is upregulated by TGF-β117,48,49; (2) enhanced intracellular degradation of procollagen prior to its secretion and extracellular deposition as fibrils47; (3) inhibition of platelet derived growth factor induced mitogenicity by interference with the activation of the mitogen activated protein kinase, a central event in the signalling cascade downstream of the activated platelet derived growth factor receptor.22
Much of the antimitogenic effect of PTX is mediated by protein kinase A which, on activation by cAMP, can phosphorylate and thus deactivate kinases upstream of mitogen activated protein kinase.50,51 In addition, PTX blocks nuclear factor κB activation in stimulated Kupffer cells which explains its activity in suppressing nuclear factor κB dependent synthesis and release of tumour necrosis factor α.52–55 However, the role of tumour necrosis factor α in fibrogenesis remains equivocal as it enhanced collagen synthesis in bleomycin induced pulmonary fibrosis56 while increasing matrix metalloproteinase expression in gut intestinal fibroblasts.57 Other antifibrotic pathways induced by PTX are independent of phosphodiesterase activity and involve second messengers such as stat-1 which is also activated by the interferons, cytokines that suppress collagen production, and fibroblast proliferation in vitro.58,59
Phosphodiesterases (PDEs) represent a family of nine isoforms (PDE1–PDE9) and several splice variants.54,55,60 These PDEs act differentially in cells, mainly due to their compartmentalisation and substrate specificity—for example, cAMP for PDE-3 and -4, and c-GMP for PDE-1 and -5. Apart from PTX which inhibits most of the PDE isoforms, and pentifylline that is approximately 10-fold more potent than PTX,17 some novel drugs block certain PDEs.54,55,60–62 Examples are sildenafil for PDE-1 and rolipram for PDE-3 that both deactivate the nuclear factor κB-tumour necrosis factor α pathway. More selective PDE inhibitors that preferentially block PDE-3 and -4 are desirable as they could potentially exert a more specific suppressive effect on activated stellate cells and myofibroblasts. Alternatively, PTX could be targeted to the fibrogenic effector cells. That this is now feasible was recently shown by use of a cyclic peptide that recognises the (activation induced) collagen VI receptor on hepatic stellate cells and myofibroblasts.63
Serum levels of a surrogate marker of hepatic fibrogenesis, PIIINP, at sacrifice, correlated with total liver collagen, histological fibrosis score, and with liver and spleen weights of fibrotic rats. This is in line with recent data that demonstrated good correlations of PIIINP with hepatic steady state levels of procollagen α1(I) and TIMP-1 mRNA levels.4 Since PIIINP, as well as a spectrum of other surrogate markers of liver fibrosis,64 can be measured in humans, prediction and even titrating of the antifibrotic effect of PTX or any other potential antifibrotic agent on a day to day basis may be possible.
In conclusion, we have shown that PTX, a phosphodiesterase inhibitor with a well known clinical profile, can dramatically downregulate hepatic procollagen I expression in biliary fibrosis. This effect is in part counteracted by upregulation of TIMP-1 which leads to a moderate net reduction in collagen accumulation. PIIINP, a surrogate marker of hepatic fibrogenesis, reflects the antifibrotic effect of PTX. More specific inhibitors for certain PDE isoforms, such as PDE-3 and -4, may be more potent antifibrotic agents. Based on these in vivo data, further studies using PDE inhibitors for treatment of hepatic fibrosis are warranted.
Acknowledgments
Supported in part by a joint grant from the German government and the University of Erlangen-Nuernberg (IZKF B18). DS was a recipient of a Hermann-und-Lilly-Schilling professorship.
Part of this work was presented in abstract form at the meeting of the American Gastroenterological Association, Orlando, Florida, May 16–19, 1999 (Gastroenterology 1999;118:A1227).