Article Text

Abstract
Background: It is now generally accepted that chronic pancreatic injury and fibrosis may result from repeated episodes of acute pancreatic necroinflammation (the necrosis-fibrosis sequence). Recent studies suggest that pancreatic stellate cells (PSCs), when activated, may play an important role in the development of pancreatic fibrosis. Factors that may influence PSC activation during pancreatic necroinflammation include cytokines known to be important in the pathogenesis of acute pancreatitis, such as tumour necrosis factor α (TNF-α), and the interleukins 1, 6, and 10 (IL-1, IL-6, and IL-10).
Aim: To determine the effects of these cytokines on PSC activation, as assessed by cell proliferation, α smooth muscle actin (α-SMA) expression, and collagen synthesis.
Methods: Cultured rat PSCs were incubated with cytokines for 24 hours. Cell proliferation was assessed by measuring 3H thymidine incorporation into cellular DNA, α-SMA expression by western blotting, and collagen synthesis by incorporation of 14C proline into collagenase sensitive protein. mRNA levels for procollagen α1(1) in PSCs were determined by northern and dot blotting methods.
Results: Expression of α-SMA by PSCs was increased on exposure to each of the cytokines used in the study. Stellate cell proliferation was stimulated by TNF-α but inhibited by IL-6, while IL-1 and IL-10 had no effect on PSC proliferation. Collagen synthesis by PSCs was stimulated by TNF-α and IL-10, inhibited in response to IL-6, and unaltered by IL-1. Changes in collagen protein synthesis in response to TNF-α, IL-10, and IL-6 were not regulated at the mRNA level in the cells.
Conclusion: This study has demonstrated that PSCs have the capacity to respond to cytokines known to be upregulated during acute pancreatitis. Persistent activation of PSCs by cytokines during acute pancreatitis may be a factor involved in the progression from acute pancreatitis to chronic pancreatic injury and fibrosis.
- pancreas
- stellate cells
- cytokines
- pancreatitis
- α-SMA, α smooth muscle actin
- GAPDH, glyceraldehyde phosphate dehydrogenase
- IL, interleukin
- PDGF, platelet derived growth factor
- PSC, pancreatic stellate cell
- SDS, sodium dodecyl sulphate
- TCA, trichloroacetic acid
- TNF-α, tumour necrosis factor α
Statistics from Altmetric.com
- α-SMA, α smooth muscle actin
- GAPDH, glyceraldehyde phosphate dehydrogenase
- IL, interleukin
- PDGF, platelet derived growth factor
- PSC, pancreatic stellate cell
- SDS, sodium dodecyl sulphate
- TCA, trichloroacetic acid
- TNF-α, tumour necrosis factor α
Pancreatitis is usually classified into two forms—acute and chronic.1 Acute pancreatitis is characterised by acute necroinflammation of the pancreas and peripancreatic tissues with, in most instances, complete recovery of pancreatic structure and function once the inflammation resolves.2 Chronic pancreatitis, on the other hand, is characterised by irreversible destruction of pancreatic structure with progressive loss of both exocrine and endocrine function.3 Traditionally, the acute and chronic forms of pancreatitis have been regarded as separate diseases.1 However, it is now being increasingly recognised (from both clinical4 and experimental studies5,6) that repeated episodes of acute pancreatitis can lead to increasing residual damage to the gland, eventually resulting in the changes of chronic pancreatitis—that is, acinar atrophy and fibrosis (the necrosis-fibrosis sequence5,7).
The mechanism(s) responsible for the development of pancreatic fibrosis has not yet been fully elucidated. However, considerable progress has been made in recent years, largely due to the identification and characterisation of stellate cells in the pancreas.8–11 Studies with these cells suggest that they may play a major role in pancreatic fibrogenesis in a manner analogous to hepatic stellate cells (the principal effector cells in liver fibrosis).12 In normal pancreas, quiescent pancreatic stellate cells (PSCs) can be identified by staining for the cytoskeletal protein desmin, a stellate cell selective marker.8 PSCs are found in a periacinar location, with long cytoplasmic processes encircling the base of pancreatic acini.8 In vitro studies with cultured PSCs have revealed that the cells store vitamin A in the form of lipid droplets in the cytoplasm, a feature similar to that described for hepatic stellate cells.8 It is hypothesised that during pancreatic injury, PSCs are activated—that is, they proliferate, transform into a myofibroblastic phenotype which exhibits positive staining for the cytoskeletal protein α smooth muscle actin (α-SMA), and synthesise and secrete increased amounts of extracellular matrix proteins, particularly collagen (such activation has been well described in hepatic stellate cells during liver injury).13 The above hypothesis is supported by results of recent studies using pancreatic sections from patients with chronic pancreatitis and from animal models of experimental pancreatic fibrosis.14,15 These studies have demonstrated the presence of activated PSCs (as identified by positive staining for α-SMA) in areas of pancreatic fibrosis. Furthermore, in situ hybridisation studies have suggested that activated PSCs are the predominant source of collagen in the fibrotic pancreas.
The factors that may activate PSCs during pancreatic injury have not yet been fully characterised although recent in vitro work has shown that alcohol (a major aetiological factor for pancreatitis) and its metabolite acetaldehyde,16 as well as oxidant stress16 and growth factors such as platelet derived growth factor (PDGF)9,17 and transforming growth factor β,9,17 can activate cultured PSCs.
Other important candidate factors that may activate PSCs during pancreatic injury are proinflammatory cytokines (such as tumour necrosis factor α (TNF-α), interleukin (IL)-1, and IL-6)18,19 known to be upregulated early in the course of acute pancreatic necroinflammation. Persistent activation of PSCs in response to repeated and/or prolonged exposure to proinflammatory cytokines (released during episodes of acute pancreatitis) may contribute to the development of pancreatic fibrosis via increased synthesis of extracellular matrix proteins, particularly fibrillar collagens (collagen types I and III). This concept would be in accordance with the necrosis-fibrosis sequence concept.
The effects of proinflammatory cytokines in vivo may be modulated by the presence of anti-inflammatory cytokines such as IL-10 (also known to be upregulated in pancreatitis).20 IL-10 is thought to be a protective cytokine and has been shown to decrease the severity of acute pancreatitis and to reduce the likelihood of systemic complications.21,22 Therefore, IL-10 may represent another putative cytokine influencing PSC activation.
The aim of this study was to examine the response of cultured PSCs to the proinflammatory cytokines TNF-α, IL-1, and IL-6, as well as the anti-inflammatory cytokine IL-10.
METHODS
Isolation and culture of PSCs
Rat PSCs were isolated as detailed previously.8 Briefly, the pancreas was digested with a mixture of collagenase P (0.05%), pronase (0.02%), and DNase (0.1%) in Gey's balanced salt solution. The resultant suspension of cells was centrifuged in a 13.2% Nycodenz gradient at 1400 g for 20 minutes. Stellate cells separated into a hazy band just above the interface of the Nycodenz solution and the aqueous buffer. This band was harvested, and the cells were washed and resuspended in Iscove's modified Dulbecco's medium containing 10% fetal bovine serum, 4 mM glutamine, and antibiotics (penicillin 100 U/ml; streptomycin 100 mg/ml). The above technique yields a preparation of stellate cells devoid of contamination by endothelial cells or macrophages, as evidenced by negative staining for the markers factor VIII and ED1, respectively.8
PSCs cultured in uncoated plastic wells (passages 1–3) were used for cytokine experiments. Quadruplicate wells of cells were exposed to increasing amounts of TNF-α (2.5–50 U/ml), IL-1 (0.001–10 ng/ml), IL-6 (0.001–10 ng/ml), or IL-10 (1–50 ng/ml) in culture medium for 24 hours at 37°C. Cells incubated with culture medium alone, without added cytokines, served as controls. Activation of PSCs was then assessed using the following indices: cell proliferation, α-SMA expression, and collagen synthesis.
Assessment of cell proliferation (3H thymidine incorporation)
Cell proliferation was estimated by measuring incorporation of 3H thymidine into trichloroacetic acid (TCA) precipitated material, as described previously.17 Briefly, cells were pulsed with 3H thymidine at a concentration of 5 μCi per well for the final six hours of the 24 hour incubation period. The reaction was stopped by aspirating the culture medium, and the cells were washed twice with Hank's balanced salt solution. Ice cold 10% TCA was added to the wells and cells incubated for 10 minutes at 4°C. This was followed by a further two washes with 5% TCA for 10 minutes, each at 4°C. NaOH 1 N (500 μl) was then added to the wells which were shaken at room temperature on a rotary shaker for 30 minutes. The reaction was stopped by adding 500 μl 1 N HCl. The mixture was transferred to scintillation vials and radioactivity of the samples was measured using a liquid scintillation counter (Tricarb 4000 Series, Canberra Packard, Five Dock, NSW, Australia).
Assessment of α-SMA expression
Levels of α-SMA in cells incubated with increasing amounts of cytokines were determined by western blotting of cell lysate proteins, using a monoclonal mouse antibody to α-SMA.17 Briefly, cells were harvested by trypsinisation, and cell lysates were obtained by incubating the cells overnight in lysis buffer (50 mM Tris HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 1% sodium dodecyl sulphate (SDS), 1 mM sodium orthovanadate, 2 mM ethylenediaminetetra acetic acid (EDTA), 10 mM NaF, 10 mg/ml aprotinin, 10 mg/ml leupeptin, and 1 mM phenylmethylsulphonyl fluoride). Samples were then centrifuged at 2200 g for 10 minutes and the supernatant was harvested for measurement of α-SMA levels.
Protein levels in cell lysates were measured by the method of Lowry and colleagues23 using bovine serum albumin as the standard. Proteins (5 μg) from each sample were separated by gel electrophoresis using a 15% SDS polyacrylamide gel. Known molecular weight protein standards were run alongside the samples. Separated proteins were transferred onto nitrocellulose membranes using a commercial semidry blotting apparatus (Biorad, Richmond, California, USA). The nitrocellulose membrane was then incubated at room temperature with 5% skim milk powder in Tris buffered saline (pH 7.6) for one hour to prevent non-specific binding of antibody. This was followed by an overnight incubation at 4°C with the primary antibody (monoclonal mouse anti-α-SMA antibody 1:200) in fresh blocking solution. The filter was then washed and incubated with the secondary antibody (goat antimouse IgG 1:1000; Amersham International, Buckinghamshire, UK) for 60 minutes at room temperature. α-SMA bands were detected by the enhanced chemiluminescence technique using the Amersham ECL kit and quantified by densitometry of scanned autoradiographs (Scion Image, Maryland, USA). Densitometry readings were expressed as integrated optical densities (arbitrary densitometer units calculated from the density as well as the size of each band) per mg protein loaded on the membranes.
Assessment of collagen synthesis
Collagen synthesis was assessed in cells incubated with increasing concentrations of cytokines by measuring the incorporation of radiolabelled proline into collagenase sensitive protein, using a modification of the method of Agelli and Wahl.24 Briefly, cells were incubated for one hour in proline free medium and then labelled for 16 hours with 2 μCi/ml 14C proline in Dulbecco's medium containing 1% fetal bovine serum, 50 mg/ml ascorbic acid, and 75 mg/ml β-aminoproprionitrile. At the end of the incubation period, culture medium in the wells was aspirated and kept aside. Cells were harvested by trypsinisation, washed, and added to the reserved medium. The samples were sonicated for 90 seconds and proteins were precipitated with 10% TCA (final concentration). TCA treated samples were centrifuged at 1000 g for 10 minutes and the pellet (comprising precipitated protein) was solubilised in 200 μl of 0.2 N NaOH. After neutralisation with 0.2 N HCl, samples were divided into two equal aliquots. One aliquot was incubated with collagenase solution (containing 6.25 mM CaCl2, 70 mM Tris (pH 7.4), 30 mM N-ethylmaleimide, and 360 U/ml collagenase type III) while the other aliquot was incubated with the above solution in the absence of collagenase for 120 minutes at 37°C. Incorporation of 14C proline into collagen and non-collagen protein was determined following precipitation with TCA. Collagen incorporated radioactivity was recovered in the TCA soluble fraction while non-collagen radioactivity was found in the TCA precipitate. Collagen production (counts per minute per ng DNA) was calculated using formulae derived by Peterkofsky and colleagues25 and was expressed as per cent of control.
Messenger RNA for procollagen α1(1)
To determine whether any observed alteration of collagen synthesis by PSCs is regulated at the transcriptional level, mRNA levels for procollagen α1(1) were assayed by northern and dot blot analysis of total cellular RNA, as described previously.16 Although the assay used for collagen protein synthesis (described above) does not identify the subtypes of collagen synthesised, previous studies on hepatic stellate cells have shown that activated stellate cells produce more collagen I than any other types of collagen (particularly III and IV). It is possible therefore that any observed change in collagen synthesis in PSCs may be regulated, at least in part, by changes in mRNA for procollagen α1(1).
Isolation of RNA
Total cellular RNA was extracted from cultured stellate cells by a modification of the method described by Chomczynski and Sacchi26 using the Tri reagent kit (Sigma Chemical Co, St Louis, Missouri, USA), as described in our previous study.16 Extracted RNA was quantified by spectrophotometry. The A260/A280 ratio of extracted RNA was routinely in the range of 1.7–1.8. Agarose gel electrophoresis of extracted RNA confirmed the integrity of the RNA samples.
Analysis of RNA
Qualitative analysis of total RNA was performed using the northern blotting technique. An RNA ladder (Sigma Chemical Co) was used for size determination. Quantitative comparisons of messenger RNA levels were made using the dot blotting technique. RNA samples (4 mg) were denatured and dot blotted in duplicate onto nylon membranes (Zeta Probe, GT blotting membrane; Biorad, California, USA). Membranes were rinsed in 2× sodium chloride sodium citrate, 0.1% SDS, sealed in plastic, and stored at −20°C until further use. cDNA probes for rat procollagen α1(1) (a kind gift from G Sparmann, University of Rostock, Germany) and for glyceraldehyde phosphate dehydrogenase (GAPDH) were used in the northern and dot blotting studies. Probes were labelled using a random priming kit (Amersham, Australia). Unincorporated nucleotides were removed using nucleic acid purification columns (Spin G50 Sephadex; Boehringer Mannheim, Germany). GAPDH expression was used as an internal control to confirm equal loading of RNA on the membranes.
Dot blots for mRNA for procollagen α1(1) and GAPDH were quantified by densitometry of autoradiographs. Washed filters were blotted dry, wrapped in plastic, and exposed to autoradiography film (Eastman Kodak Company, New York, USA). The autoradiographs were subjected to scanning densitometry. Readings were calculated as integrated optical density units (derived from the density as well as the size of each band/dot) per μg RNA loaded on the membranes and expressed as per cent of control.
Protein and DNA determinations
Protein content of cell lysates was measured by the method of Lowry and colleagues23 using bovine serum albumin as the standard. Pancreatic DNA was assayed by a modified fluorimetric microassay as described by Kapuscinski and Skoczylas27 using calf thymus DNA as the standard.
Statistical analysis
Results are expressed as means (SEM) for five separate cell preparations per experimental protocol. Data were analysed using repeated measures ANOVA.28 Fisher's protected least significant difference was used for comparison of individual groups provided the F test was significant.28,29 The analyses were performed using the Statview II statistical software package.29
Materials
All general chemicals were of analytical reagent grade and were purchased from Sigma Chemical Co. (St Louis, Missouri, USA). Collagenase P was purchased from Boehringer Mannheim (Mannheim, Germany), collagenase type III (from Clostridium histolyticum), and protease type XIV (from Streptomyces griseus) were obtained from the Sigma Chemical Co. and DNase was purchased from Pharmacia Biotech (Uppsala, Sweden). Cell culture reagents were purchased from Sigma Chemical Co. Nycodenz was obtained from Nycomed Pharma AS, Oslo, Norway. Iscove's modified Dulbecco's medium was purchased from Gibco BRL. Antibodies were obtained from the following sources: monoclonal antibody to α-SMA (Sigma Chemical Co.), goat antimouse horseradish peroxidase conjugated antibody (Dako Corporation, Carpintaria, California, USA). 3H thymidine (specific activity 6.7 Ci/mmol) was purchased from ICN Pharmaceuticals (Costa Mesa, California, USA) while 14C proline (specific activity 250 Ci/mmol) was obtained from NEN (Boston, Massachusetts, USA). Rat recombinant TNF-α, IL-6, and IL-10, and murine recombinant IL-1 were purchased from Peprotech (Rocky Hill, New Jersey, USA).
RESULTS
α-SMA expression in response to cytokines
Immunoblotting for α-SMA revealed a single band in each lane corresponding to the known molecular weight of α-SMA (42 kDa). Densitometry of autoradiographs showed that TNF-α, at concentrations of 2.5 and 5 U/ml, caused a trend towards increased α-SMA expression. At a TNF-α concentration of 10 U/ml the increase in α-SMA expression achieved statistical significance: 121 (3.1)% of control (p<0.05) (fig 1A). However, with higher concentrations of TNF-α (25 and 50 U/ml), α-SMA expression fell to baseline values. The reasons for the return of α-SMA to baseline expression with high TNF-α concentrations are not immediately apparent. IL-1, at concentrations of 0.001, 0.01, and 0.1 ng/ml, also stimulated significant increases in α-SMA expression to 690 (179)%, 954 (423)%, and 907 (361)% of control, respectively (p<0.01) (fig 1B). IL-6 caused a biphasic increase in α-SMA expression in PSCs, with peaks at 0.001 ng/ml (175 (28)%; p<0.05) and 10 ng/ml (184 (75)%; p<0.05) (fig 1C). α-SMA expression by PSCs was increased by exposure to 10 ng/ml IL-10 (289 (127)% of control; p<0.05) (fig 1D).

Effect of cytokines on α-smooth muscle actin (α-SMA) expression by pancreatic stellate cells (PSCs). Data are expressed as per cent of control (mean (SEM); n=5 cell preparations). (A) Tumour necrosis factor α (TNF-α, at a concentration of 10 U/ml) significantly increased α-SMA expression in PSCs. (B) Interleukin (IL)-1 (0.001, 0.01, and 0.1 ng/ml) significantly increased α-SMA expression in PSCs. (C) IL-6 caused a biphasic increase in α-SMA expression in PSCs, with peaks at 0.001 ng/ml and 10 ng/ml. (D) IL-10 (10 ng/ml) also increased expression of α-SMA in PSCs. MW, molecular weight.
Cell proliferation in response to cytokines
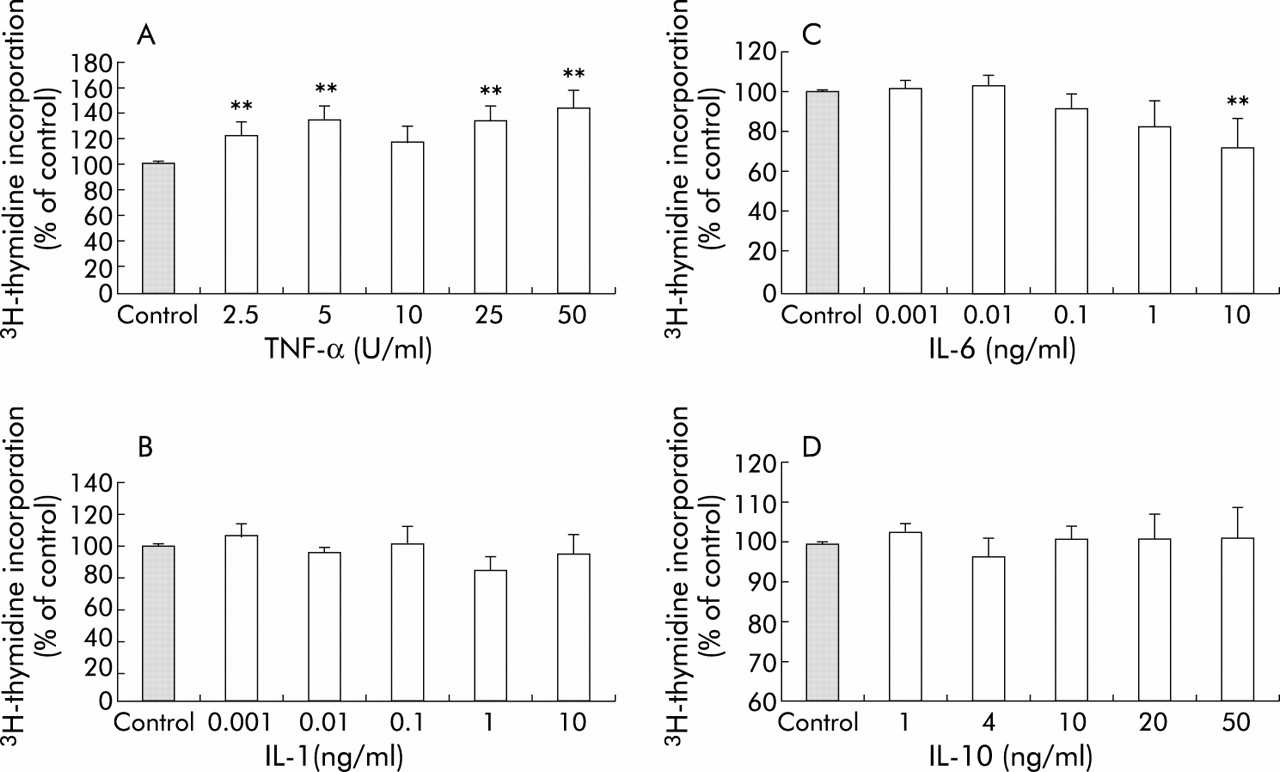
Exposure to TNF-α (50 U/ml) increased the rate of proliferation of PSCs, as indicated by a statistically significant increase in incorporation of 3H thymidine to 144 (13)% of control levels (p<0.01) (fig 2A). PSC proliferation was unaffected by exposure to IL-1 (fig 2B). IL-6 at the highest concentration used (10 ng/ml) inhibited PSC proliferation, as indicated by a decrease in 3H thymidine incorporation to 72 (14)% of control (p<0.01) (fig 2C). As with IL-1, IL-10 had no effect on PSC proliferation (fig 2D).
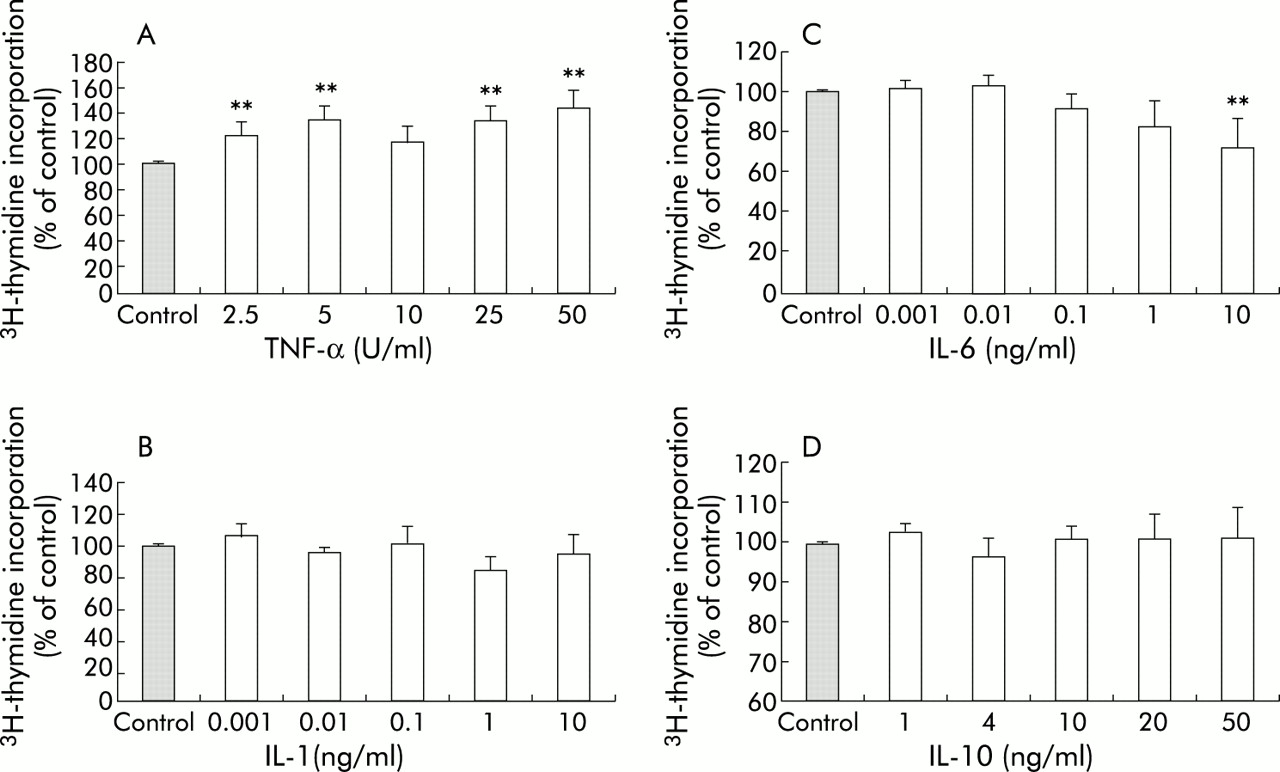
Effect of cytokines on DNA synthesis in pancreatic stellate cells (PSCs). Data are expressed as per cent of control (mean (SEM); n=5 separate cell preparations). (A) Tumour necrosis factor α (TNF-α) significantly increased DNA synthesis in PSCs to a maximum of 144 (13)% of control levels. (B) Interleukin (IL)-1 had no effect on DNA synthesis in PSCs. (C) IL-6, at a concentration of 10 ng/ml, inhibited DNA synthesis in PSCs, as demonstrated by a decrease in 3H thymidine incorporation to 72 (14)% of control. (D) IL-10 had no effect on DNA synthesis in PSCs.
Collagen synthesis and procollagen α1(1) mRNA expression in response to cytokines
Collagen protein synthesis was measured as counts per minute of 14C proline incorporated into collagenase sensitive protein per ng DNA and the results are expressed as per cent of control levels. PSCs incubated with TNF-α showed increased collagen protein synthesis compared with control cells (fig 3A). This effect was greatest at a concentration of 10 U/ml TNF-α (203 (45)%; p<0.05). IL-1 did not have any effect on collagen protein synthesis by PSCs (fig 3B). Similarly, collagen synthesis was unaffected by IL-6, except at the highest concentration of the cytokine (10 ng/ml) when collagen synthesis was found to be significantly lower than control levels (49.7 (7.8)% of control; p<0.01) (fig 3C). Collagen protein synthesis by PSCs was significantly increased (203.9 (40.6)% of control; p<0.05) by exposure to the anti-inflammatory cytokine IL-10 at the highest concentration used—that is, 50 ng/ml (fig 3D).

Effect of cytokines on collagen synthesis by pancreatic stellate cells (PSCs). Data are expressed as per cent of control (mean (SEM); n=5 separate cell preparations). (A) Tumour necrosis factor α (TNF-α, 10 U/ml) significantly increased collagen synthesis. (B) PSCs incubated with interleukin (IL)-1 showed no change in collagen synthesis. (C) PSCs incubated with 10 ng/ml IL-6 exhibited a decrease in collagen synthesis to 49.7 (7.8)% of control levels. (D) IL-10, at a concentration of 50 ng/ml, stimulated collagen synthesis by PSCs to 203 (40)% of control levels.
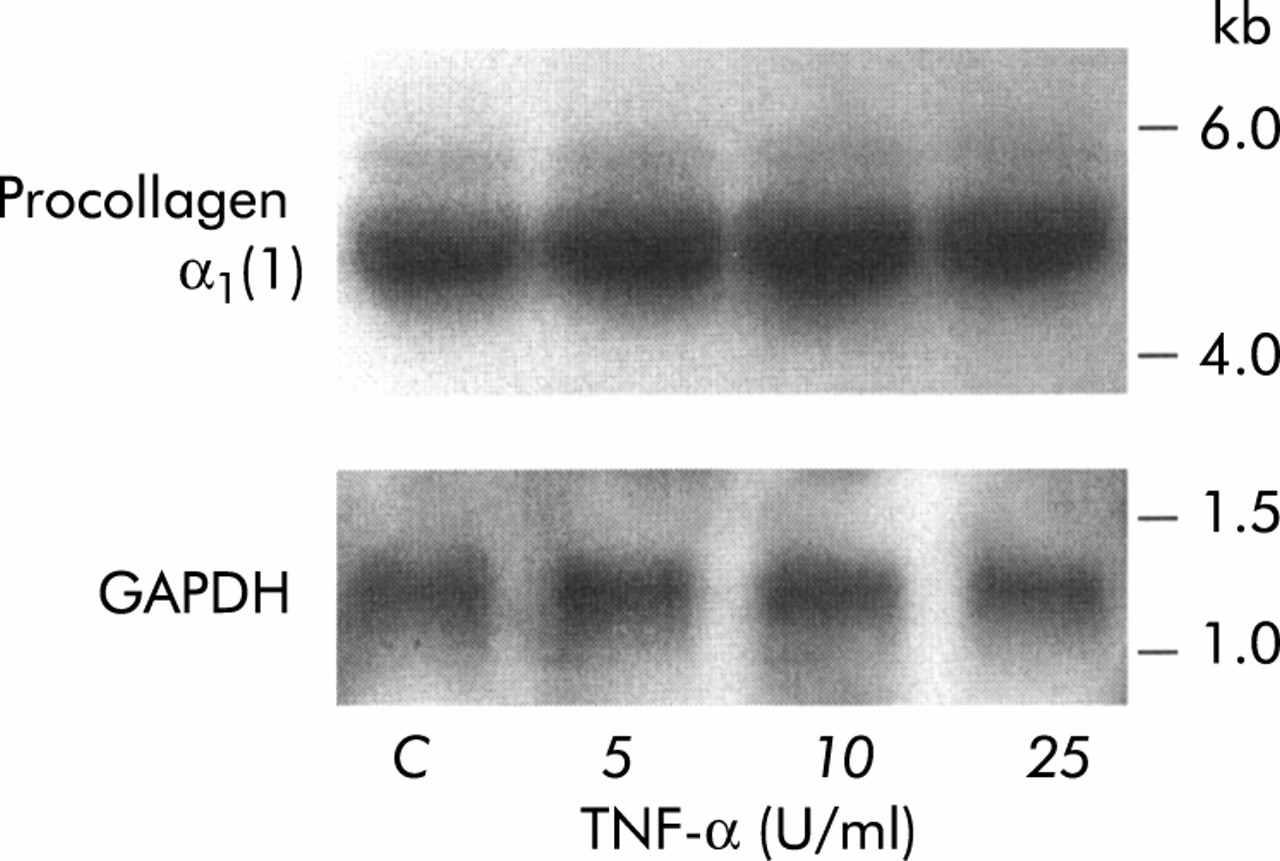
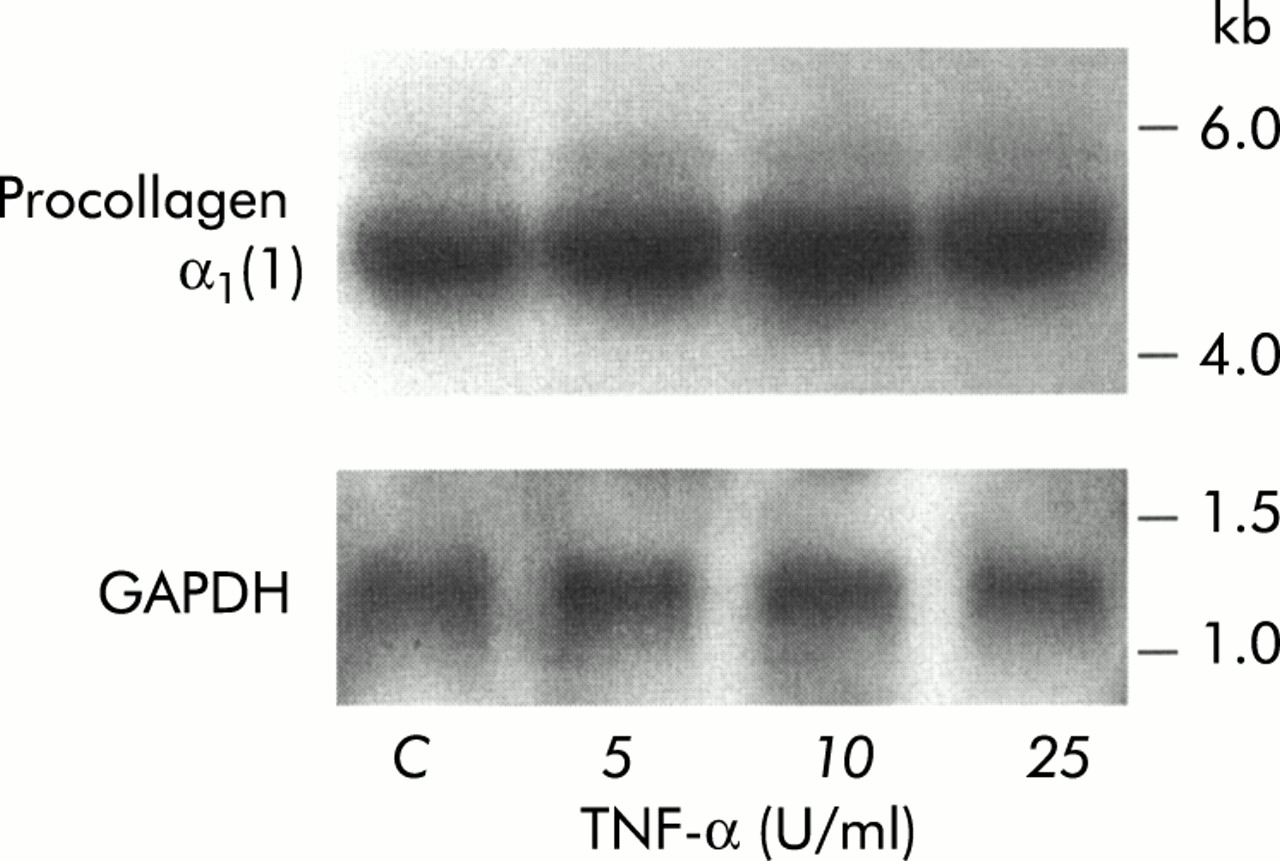
To determine whether the alterations in collagen protein synthesis induced in PSCs by TNF-α, IL-6, and IL-10 were regulated at the mRNA level in cells, procollagen α1(1) mRNA expression was assessed in PSCs exposed to these cytokines. Northern hybridisation revealed that mRNA for procollagen α1(1) was of the expected size (5.4 kb). Figure 4 depicts a representative northern blot for procollagen α1(1) and GAPDH in TNF-α-treated PSCs. Results of quantitative analysis (scanning densitometry) of dot blots for procollagen α1(1) mRNA in PSCs treated with TNF-α, IL-6, and IL-10 are presented in table 1. No difference between procollagen α1(1) levels in control cells and cytokine treated cells was observed, suggesting that the observed increase in collagen protein synthesis was regulated at a post-transcriptional level in the cells.
Densitometry of dot blots for procollagen-α1(1)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative northern blot showing procollagen-α1(1) and glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA expression in pancreatic stellate cells treated with tumour necrosis factor α (TNF-α).
DISCUSSION
This study has demonstrated that PSCs are activated on exposure to the proinflammatory cytokines TNF-α, IL-1, and IL-6 and also when exposed to the anti-inflammatory cytokine IL-10. While each of the above cytokines increased α-SMA expression in PSCs, their effects on the other two activation parameters examined (cell proliferation and collagen synthesis) varied. Of the proinflammatory cytokines, TNF-α increased both cell proliferation and collagen synthesis, IL-6 decreased cell proliferation and collagen synthesis, while IL-1 had no effect on these two parameters. The anti-inflammatory cytokine IL-10 did not influence cell proliferation but increased collagen synthesis by PSCs.
It should be noted that the PSCs used in this study were preactivated by culture on uncoated plastic. The observation that these cells can be activated even further by exposure to cytokines may have important implications for the situation in vivo. Our findings suggest that PSCs activated during pancreatic injury by cellular stressors such as oxidant stress may be capable of further activation by cytokines released during pancreatic necroinflammation. Thus it is possible that a number of factors exert synergistic effects on PSCs in vivo leading to a perpetuation of their activated state.
All proinflammatory cytokines examined in this study (TNF-α, IL-1, IL-6) have been shown to be produced within the pancreas during the early stages of acute pancreatitis and are found in elevated concentrations in both blood and pancreatic tissue.30 A correlation exists between serum levels of these cytokines and disease severity.31–36 In particular, serum levels of IL-6 are now regarded as a reliable clinical indicator of the severity of acute pancreatitis.32,33 The major sources of TNF-α, IL-1, and IL-6 during pancreatic necroinflammation are activated macrophages in the inflammatory infiltrate within the gland. In addition, pancreatic acinar cells have been found to express these cytokines during experimental acute pancreatitis.18,37 Endogenous expression of the above cytokines by PSCs has not yet been established.
TNF-α was found to influence each of the three parameters of PSC activation examined in this study, namely α-SMA expression, cell proliferation, and collagen synthesis. The effect of TNF-α on α-SMA expression concurs with the findings reported with hepatic stellate cells.38 The observed mitogenic effect of TNF-α on PSCs in vitro represents an important finding. Recent in vivo studies using animal models of pancreatic necroinflammation and fibrosis have reported an increase in the number of PSCs (as indicated by increased staining for the stellate cell selective marker desmin) in fibrotic areas of the pancreas.14 Given the results of this study, it is possible that TNF-α, either alone or in conjunction with other known stellate cell mitogens such as PDGF,17 mediates stellate cell proliferation under conditions of pancreatic injury.
TNF-α was also found to stimulate total collagen protein synthesis in PSCs. The predominant collagen isotype in normal healthy tissue is collagen type IV whereas in fibrosis there is accumulation of fibrillar collagens, particularly collagen type I. In the present study, it was found that TNF-α had no effect on mRNA levels for procollagen α1(1) (which codes for collagen type I), suggesting that the TNF-α induced increase in collagen protein synthesis (at least with respect to the collagen type I component) is regulated at a post-transcriptional level in PSCs. However, it has to be acknowledged that mRNA for other components of fibrillar collagens that were not examined in this study (for example, collagen type III) may be influenced by cytokine treatment. To the best of our knowledge, there are no studies in the published literature examining the effects of TNF-α on collagen synthesis by PSCs. In contrast, the response of hepatic stellate cells to TNF-α has been the subject of a number of studies and investigators have variously reported an increase, decrease, or no change in collagen synthesis by these cells.39–41 Variations in experimental conditions or methodology, or variations in the collagen isotype studied may explain the disparate findings.
IL-1 increased α-SMA expression but did not alter the rate of cell proliferation or collagen synthesis in cultured PSCs. This cytokine has a spectrum of biological activities similar to TNF-α42,43 and is thought to play a role in the pathogenesis of acute pancreatitis. The effects of IL-1 on PSCs have not been reported previously. With respect to the effects of this cytokine on hepatic stellate cells, both stimulation and inhibition of cell proliferation and collagen synthesis have been documented,38,44,45 but α-SMA expression in response to IL-1 has not yet been studied. The observed increase in α-SMA expression in PSCs on exposure to IL-1 suggests that IL-1 may be a mediator of stellate cell activation during pancreatic injury in vivo.
In the present study, IL-6 was found to increase α-SMA expression in PSCs. In contrast, cell proliferation and collagen synthesis were significantly reduced by IL-6 (albeit only at the highest concentration used—10 ng/ml). The effects of IL-6 on α-SMA expression or cell proliferation have not been examined previously in stellate cells from either the pancreas or the liver. With respect to collagen synthesis, an increase has been reported in hepatic stellate cells exposed to concentrations of up to 20 ng/ml.46,47 However, higher concentrations of the cytokine (40 ng/ml) have been reported to inhibit collagen synthesis in these cells. The reasons for the differences in collagen synthesis in pancreatic and hepatic stellate cells in response to IL-6 are not immediately apparent although it should be noted that studies with hepatic stellate cells used human IL-6 whereas the present study used rat IL-6.
IL-6, in addition to its proinflammatory effects, is known to have many anti-inflammatory effects (including downregulation of TNF-α and IL-1, and induction of adrenocorticotrophic hormone and glucocorticoid synthesis). In vivo, IL-6 is produced in response to increased TNF-α and IL-1 secretion in many cell types. Indeed, hepatic stellate cells have been shown to synthesise IL-6 when activated by TNF-α, IL-1, or bacterial endotoxin.48 The possibility of IL-6 production by PSCs has not yet been examined. Nevertheless, the findings of this study suggest that IL-6, whatever its source, has the potential to act as a regulatory factor for the activation of PSCs during pancreatic injury in vivo.
IL-10, a predominantly anti-inflammatory cytokine, is known to be upregulated in acute pancreatitis.20 IL-10 inhibits the synthesis of proinflammatory cytokines, including TNF-α, IL-1, IL-6, and IL-8 by monocytes and macrophages and is thought to play a protective role in acute pancreatitis. The anti-inflammatory function of IL-10 has been confirmed by a number of in vivo studies using IL-10 deficient animals.49,50 Acute inflammation of the pancreas and liver is reported to be more severe in IL-10 knockout animals49,50 while exogenous IL-10 has been shown to decrease the severity of experimental acute pancreatitis.21,51 The results of the present study demonstrating that exogenous IL-10 activated PSCs were unexpected. There are no published studies to date examining the effects of exogenous IL-10 on stellate cells in the liver or pancreas. IL-10 has been reported to be produced endogenously in activated hepatic stellate cells.52 It is postulated that IL-10 may play a regulatory role in the response of these cells to activating factors such as TNF-α or endotoxin. Using neutralising antibodies to block endogenous IL-10 activity, it has been shown that IL-10 downregulates collagen synthesis in both unstimulated and activated hepatic stellate cells.52 These findings are in contrast with the IL-10 induced increase in collagen synthesis observed in the present study. The different results may be related to the use of exogenous IL-10 in the present study. Endogenous production of IL-10 by PSCs has not yet been studied.
The present study has examined the direct effects of individual cytokines on PSCs in vitro (results summarised in table 2). It has to be acknowledged however that during necroinflammation in vivo, multiple cytokines and other inflammatory mediators are released simultaneously within the gland. These factors may have synergistic, antagonistic, or complementary effects on PSCs. For example, it has been reported that while TNF-α or IL-1 alone have a mitogenic effect on fibroblasts, the combination is antiproliferative.53 Similarly, in the presence of IL-10, activation of hepatic stellate cells by TNF-α is inhibited.52 Thus the ultimate response of PSCs during pancreatic necroinflammation in vivo may be modulated by the combination of cytokines present at the time within the gland.
Summary of results
In summary, this study is the first to demonstrate that PSCs have the capacity to respond to cytokines known to be upregulated in acute pancreatitis. As such, PSCs have the potential to be actively involved in both the recovery phase as well as the progression of the disease. For example, during a self limiting attack of acute pancreatitis, cytokine activated PSCs may participate in tissue repair by regulating extracellular matrix deposition in the gland. On the other hand, during repeated attacks of pancreatitis and consequent repeated or continued exposure to cytokines, PSCs may attain a persistently activated state, thereby leading to pathological increases in collagen synthesis, eventually resulting in the development of pancreatic fibrosis.
Acknowledgments
This project was supported by grants from the National Health and Medical Research Council of Australia and the Australian Brewers' Foundation.
REFERENCES
Linked Articles
- Editor's quiz: GI snapshot
- PostScript
- Editor's quiz: GI snapshot
- Editor's quiz: GI snapshot