Article Text

Abstract
Background: Mutations in the PRSS1 gene explain most occurrences of hereditary pancreatitis (HP) but many HP families have no PRSS1 mutation. Recently, an association between the mutation N34S in the pancreatic secretory trypsin inhibitor (SPINK1 or PSTI) gene and idiopathic chronic pancreatitis (ICP) was reported. It is unclear whether the N34S mutation is a cause of pancreatitis per se, whether it modifies the disease, or whether it is a marker of the disease.
Patients and methods: A total of 327 individuals from 217 families affected by pancreatitis were tested: 152 from families with HP, 108 from families with ICP, and 67 with alcohol related CP (ACP). Seven patients with ICP had a family history of pancreatitis but no evidence of autosomal dominant disease (f-ICP) compared with 87 patients with true ICP (t-ICP). Two hundred controls were also tested for the N34S mutation. The findings were related to clinical outcome.
Results: The N34S mutation was carried by five controls (2.5%; allele frequency 1.25%), 11/87 (13%) t-ICP patients (p=0.0013 v controls), and 6/7 (86%) affected (p<0.0001 v controls) and 1/9 (11%) unaffected f-ICP cases. N34S was found in 4/108 affected HP patients (p=0.724 v controls), in 3/27 (11%) with wild-type and in 1/81 (1%) with mutant PRSS1, and 4/67 ACP patients (all p>0.05 v controls). The presence of the N34S mutation was not associated with early disease onset or disease severity.
Conclusions: The prevalence of the N34S mutation was increased in patients with ICP and was greatest in f-ICP cases. Segregation of the N34S mutation in families with pancreatitis is unexplained and points to a complex association between N34S and another putative pancreatitis related gene.
- chronic pancreatitis
- idiopathic pancreatitis
- alcohol
- SPINK1
- PSTI
- HP, hereditary pancreatitis
- ICP, idiopathic chronic pancreatitis
- f-ICP, familial ICP
- t-ICP, true ICP
- ACP, alcohol related chronic pancreatitis
- PCR, polymerase chain reaction
- RFLP, restriction fragment length polymorphism
- BSA, bovine serum albumin
- CFTR, cystic fibrosis transmembrane conductance regulator
- EUROPAC, European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer
Statistics from Altmetric.com
- HP, hereditary pancreatitis
- ICP, idiopathic chronic pancreatitis
- f-ICP, familial ICP
- t-ICP, true ICP
- ACP, alcohol related chronic pancreatitis
- PCR, polymerase chain reaction
- RFLP, restriction fragment length polymorphism
- BSA, bovine serum albumin
- CFTR, cystic fibrosis transmembrane conductance regulator
- EUROPAC, European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer
Hereditary pancreatitis (HP) is characterised by recurrent attacks of painful acute pancreatitis from an early age eventually resulting in chronic pancreatitis, with loss of pancreatic exocrine and endocrine function.1–3 HP is an autosomal dominant condition with a penetrance of approximately 80%.1,4 Causative mutations have been identified in the PRSS1 (PRoteaSe Serine 1) gene which encodes cationic trypsinogen.5–7 The two mutations most frequently identified are the N29I mutation, which may indirectly increase the frequency of autoactivation,8 and the R122H mutation, which is thought to alter a trypsin sensitive hydrolysis site and so stabilise trypsin once activated.9,10 A third less common mutation (A16V) with a lower penetrance lies just beyond the signal peptide and may affect trypsinogen transportation.11 Other rare PRSS1 mutations include R122C,12 N29T,12 D22G,13 and K23R,14 all of which are associated with HP.
Many families with a high frequency of pancreatitis, consistent with an autosomal mode of inheritance, carry no mutation in PRSS1.7,11 Approximately one third of all patients have no aetiological factor contributing to the development of chronic pancreatitis and are said to have idiopathic chronic pancreatitis (ICP).15 Witt and colleagues16 recently reported a strong association between mutations in a gene encoding the serine protease inhibitor, Kazal type 1 (SPINK1; also known as pancreatic secretory trypsin inhibitor, PSTI; OMIM 167790) and ICP. The human gene has four exons and is located on chromosome 5.17 The mature peptide consists of 56 amino acids and has a reactive site at position 18 that is a specific target substrate for trypsin.18 SPINK1 inhibits approximately 20% of total trypsin activity within the pancreas, thus providing an important defence against prematurely activated trypsinogen.19 Previously, Chen and colleagues20 investigated the possible link between the SPINK1 gene and HP and found an A→G transition causing an N34S (also referred to as N11S20) substitution in one HP family that did not carry any of the PRSS1 mutations. The mutation was found not to segregate with the disease and was therefore excluded as a potential disease causing mutation.
Witt and colleagues16 found that the N34S mutation was present in 18 (21%; six homozygotes and 12 heterozygotes) of 85 patients who did not carry any of the PRSS1 mutations. They further reported a strong association between the SPINK1 locus and pancreatitis in families with individuals carrying a mutation (N34S) in SPINK1 using the transmission disequilibrium test and concluded that there was a causative link. Pfützer and colleagues21 then performed genetic linkage analysis on five HP families and excluded significant linkage between pancreatitis and the SPINK1 locus (5q31.1-2). The SPINK1 gene of 112 PRSS1 mutation negative patients was sequenced and three novel mutations and intronic polymorphisms were identified. The N34S mutation was observed in five (9.1%) of 55 patients with HP and in 23 (40.4%; seven homozygotes, 14 heterozygotes, and two compound heterozygotes) of 57 patients with ICP.
Thus whether an N34S mutation should be considered a causative factor of pancreatitis per se or simply a disease modifier is uncertain.22–24 Multiple SPINK1 sequence variants other than N34S have been identified but their clinical significance is even less certain.16,21 To investigate this further, large groups of patients with HP, ICP, and alcohol related chronic pancreatitis (ACP) were tested for the N34S mutation.
METHODS
Patients
Patients and family members with HP and some individuals with ICP were recruited through the European Registry of Hereditary Pancreatitis and Familial Pancreatic Cancer (EUROPAC). Other patients with ICP and all patients with ACP were recruited from the Pancreas Unit based at the Royal Liverpool University Hospital, Liverpool, UK.
The diagnosis of chronic pancreatitis was based on clinical and radiological criteria and exocrine pancreatic function tests.25 Following written informed consent both affected individuals and their referring clinicians completed detailed questionnaires relating to clinical history, radiology, pancreatic function tests, interventions, and histology. Clinically unaffected individuals from relevant families were rigorously screened for symptoms of pancreatitis by questionnaire. The diagnosis of diabetes mellitus was based on standard tests and/or the requirement for insulin, and the diagnosis of exocrine failure was based on standard diagnostic tests and/or the use of pancreatic enzyme supplements. ACP was diagnosed in a patient with chronic pancreatitis and an alcohol consumption of ≥80 g/day.25 ICP was defined as chronic pancreatitis occurring in an individual for which there was no known cause after a detailed history, biochemical testing, and radiology, including that of both the biliary and pancreatic ducts, and in the absence of other affected individuals within that family.25 A diagnosis of HP was made on the basis of two first degree relatives or three or more second degree relatives, in two or more generations with recurrent acute pancreatitis, and/or chronic pancreatitis for which there were no precipitating factors. Cases in which the criteria for HP were not met, yet in whom there was a familial trend (mainly in cases in which there was more than one affected individual within the same generation), were subclassified as having familial ICP (f-ICP). The other subgroup were those with the true form of ICP (t-ICP) in which there was only one known family member affected. A multidisciplinary professionally experienced committee accepted or rejected individuals and families onto the register and agreed on the relevant diagnostic category after consideration of all of the relevant information.
At the censor date, EUROPAC had studied 152 individuals from 59 families with HP (tables 1, 2). There were 108 affected patients (from 59 families), of whom 81 patients from 45 families had one of three PRSS1 mutations (R122H=53; N29I=23; A16V=5) and 27 patients from 14 families with wild-type PRSS1. In addition, there were 44 unaffected individuals from 27 HP families (21 families characterised by mutant and six by wild-type PRSS1 in affected individuals). The N34S mutation was also tested in 94 patients with ICP (t-ICP=87; f-ICP=7) and in 14 unaffected relatives from four of their families (nine from one f-ICP family and five from three t-ICP families). Sixty seven patients with ACP were also tested. Control DNA samples for N34S analysis were obtained from 200 randomised, anonymous, healthy blood donors from Liverpool, UK (100) and Münster, Germany (100).
Clinical data collected included age at symptom onset, age at pancreatic endocrine and exocrine insufficiency, duration of attacks, frequency of attacks, number of hospital admissions, surgery for pancreatic complications, and age at operation for pancreatic complications.
This study was undertaken under strict ethical guidelines and involved written informed consent of every participant.
Mutation detection
DNA extraction
All blood and DNA specimens were processed in the EUROPAC Laboratories at the Merseyside Regional Molecular Genetics Laboratories, Liverpool Women's Hospital and the Department of Surgery, Royal Liverpool University Hospital, Liverpool, UK. Genomic DNA was extracted from peripheral blood leucocytes according to standard protocols (Qiagen, West Sussex, UK).
PRSS1 mutation detection
Patients with HP and ICP were screened for the common mutations of PRSS1. The R122H mutation was detected using the method described by Howes and colleagues.26 The N29I mutation was detected by amplification of a region of exon 2 of PRSS1 using the primers 5`-CCATCTTACCCAACCTCAGTAG-3` and 5`-TGATGACAGATCGTTGGGGGCTAGA-3`; a restriction site for Sau3A is produced in the presence of the N29I mutation. Digestion with Sau3A cleaves 23 base pairs from the amplified product only if the mutation is present. Polymerase chain reaction (PCR) amplification was followed by agarose gel electrophoresis of the PCR products using a 1.5% (w/v) agarose gel (Synergel; New England Bio Labs. Hitchin, Hertfordshire, UK). The A16V mutation was detected using a PCR-restriction fragment length polymorphism (RFLP) technique based on the elimination of the Fnu4HI restriction site by the mutation, as described by Witt and colleagues.11
Where there was evidence of a familial association with chronic pancreatitis (f-ICP or HP) but where there was no recognised mutation, all five exons of the PRSS1 gene were sequenced in both directions5 using the ABI 377 automated fluorescent sequencer (Applied Biosystems; Perkin Elmer, Warrington, UK) and the Big Dye terminator sequencing kit (Applied Biosystems; Perkin Elmer).
N34S mutation detection
Restriction fragment length polymorphism (RFLP) analysis
Primers were designed to amplify exon 3 based on the published nucleotide sequence (GenBank, NM-003122). The forward and reverse primer sequences were 5`-TTCTGTTTAATTCCATTTTTAGGCCAAATGCTGCA-3` and 5`-GGCTTTTATCATACAAGTGACTTCT-3`, respectively. The primers were designed to introduce a PstI endonuclease restriction site in sequences containing the N34S mutation and a BsrDI endonuclease restriction site in wild-type sequences. A polymerase with 3` to 5` proof reading activity was chosen to eliminate PCR error. PCR was performed using 50 ng genomic DNA template, 0.5 U Pfu DNA polymerase (Promega, Southampton, UK), 2 mmol/l MgCl2, 200 μmol/l of each deoxynucleotide triphosphate (Roche Diagnostics, East Sussex, UK), and 200 nmol/l of each primer in a 50 μl reaction volume. Thirty five cycles of PCR were performed (30 seconds at 94°C, 30 seconds at 60°C, and 60 seconds at 72°C). The PCR products were then digested with restriction endonucleases PstI and BsrDI. PCR product (25 μl) was added to 10 U PstI (New England Biolabs), 1× digest buffer (New England Biolabs), and 1× bovine serum albumin (BSA) (New England Biolabs) to a final volume of 50 μl and incubated at 37°C for one hour. The remaining 25 μl of PCR product was added to 10 U BsrDI (New England Biolabs), 1× digest buffer (New England Biolabs), 1× BSA (New England Biolabs) to a final volume of 50 μl and incubated at 55°C for one hour. The digestion reactions were heat inactivated by incubation at 80°C for 15 minutes. The products were analysed by agarose gel electrophoresis using a 3% (w/v) Nusieve 3:1 agarose (Flowgen, Leicestershire, UK) gel, 1× TAE buffer, and 0.5 μg/ml ethidium bromide. Undigested amplification products were 320 bp in length. After digestion with PstI a product of 286 bp was obtained from mutant sequences and an identical result was achieved from wild-type sequences after digestion with BsrDI. Heterozygote samples produced both products of 320 bp and 286 bp after digestion with either endonuclease (fig 1). To validate the RFLP analysis further, the PCR reactions were repeated and the amplification products purified by ethanol precipitation prior to digestion. The digestion reactions were then incubated for 16 hours. The results obtained were identical in both experiments.
Sequence analysis
Primers were designed from intronic sequences flanking exon 3, based on the published nucleotide sequence. The forward and reverse primer sequences were 5`-AATAGCAGAGGCATG ACTTA-3` and 5`-AATCCAAGCTATCGACTATT-3`, respectively (designed from GenBank AF286028). PCR was performed using 50 ng genomic DNA template, 0.5 U Pfu DNA polymerase (Promega), 2 mmol/l MgCl2, 200 μmol/l each deoxynucleotide triphosphate (Roche Diagnostics), and 200 nmol/l of each primer in a 50 μl reaction volume. Thirty five cycles of PCR were performed (30 seconds at 94°C, 30 seconds at 60°C, 60 seconds at 72°C). Cycle sequencing was performed on the products directly using the ABI Prism Big Dye Terminator Cycle Sequencing kit (PE Biosystems). Sequencing reactions consisted of 100 ng template, 3.2 pmol primer, 4 μl of Cycle Sequencing premix, and 4 μl of sequencing buffer in a total volume of 20 μl. Reaction products were purified by isopropanol precipitation and run on an ABI Prism 377 DNA Analyser (PE Biosystems).
Statistical analysis
All statistical analysis was carried out using the software Statview 5 (SAS Institute Inc., Cary, North Carolina, USA, 1998). Categorical data were analysed by Fisher's exact probability test. The principal contingency analyses were the prevalence of the N34S mutation in patients with ICP, HP, and ACP compared with controls; the remaining analyses were performed for consistency. Continuous data were analysed by the Kruskall-Wallis and Mann-Whitney U tests and interactions by analysis of variance (ANOVA). The cumulative incidence was calculated by the Kaplan-Meier method and statistical comparisons were made by Weibull or log rank (Mantell-Cox) analysis. Significance was set at p<0.0025 (Bonferroni correction).
RESULTS
N34S mutation screening technique
The RFLP screening technique detected 31 individuals with the N34S mutation (heterozygous in 29 cases and homozygous in two cases) and was confirmed by direct sequencing in each case. In addition, 80 cases with no N34S mutation by the RFLP technique were shown to have wild-type sequences in exon 3 of the SPINK1 gene by direct sequencing. Thus the PstI and BsrDI restriction digests were shown to identify the presence or confirm the absence of the N34S mutations with 100% accuracy.
Frequency of the N34S mutations in the study groups
The frequencies of the N34S mutation in the different groups are summarised in tables 2–4 and the results of contingency analysis are given in table 5. Four of 100 (4%) blood donors from Liverpool, UK, and one of 100 (1%) blood donors from Münster, Germany, carried the N34S mutation and all were heterozygous. This gave an average prevalence of the N34S mutation among controls of 2.5% and an allele frequency of 1.25%.
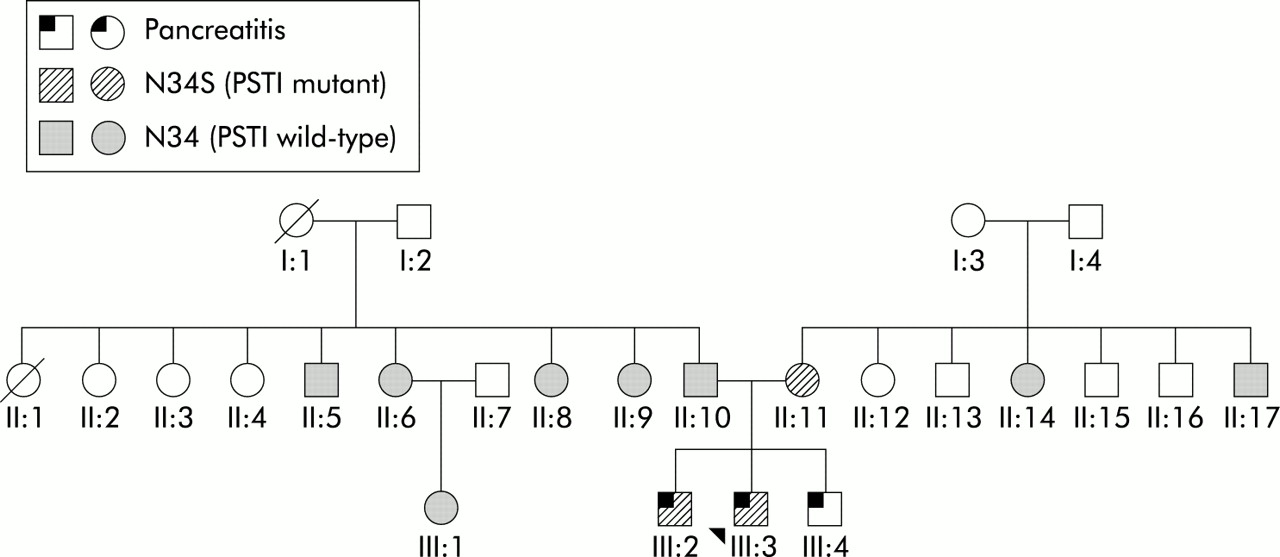
Seventeen (18%) of 94 patients with ICP carried the N34S mutation (p<0.0001 v controls), two of whom were homozygous for the mutation. No other cases of homozygosity for N34S were identified in our cohort. Of the ICP patients, 11 (13%) of 87 patients with t-ICP carried the N34S mutation (p=0.0013 v controls), including two patients homozygous for the mutation. Six (86%) of seven affected individuals with f-ICP carried N34S, at least one of which was from each of the four families. An affected individual with no N34S mutation was also found in two of the four f-ICP families. In one family, one (11.1%) of nine unaffected individuals also carried the mutation but was the mother of three affected brothers (fig 2). Five unaffected relatives of three patients with t-ICP all tested negative.
In families with HP, only four (4%) of 108 affected patients carried the N34S mutation (p=0.724 v controls). Patients with HP and wild-type PRSS1 had a higher prevalence of N34S than HP patients with PRSS1 mutations but the difference was only of marginal statistical significance (3/27 wild-type PRSS1 patients compared with 1/81 patients with a PRSS1 mutation; p=0.047). Furthermore, the prevalence of N34S in HP patients with no PRSS1 mutation grouped with f-ICP patients was significantly higher than in controls (9/34 compared with 5/200; p<0.0001).
Ten individuals from families with wild-type PRSS1 carried the SPINK1 mutation. Two of these were affected homozygotic twins; as no other family member had pancreatitis, including both parents, these were considered as f-ICP. Two families (one with HP and one with f-ICP) had two affected individuals, both with N34S. The latter, f-ICP family (fig 2), has been described above. The remaining three patients came from separate families, which in two families (one HP and one f-ICP) included another affected family member who did not have the N34S mutation.
Only one (1.9%) of 53 patients with the R122H mutation in PRSS1 had the N34S mutation. None of the 28 patients with other PRSS1 mutations carried the N34S SPINK1 mutation (N29I=23; A16V=5). Only one of 58 unaffected relatives of patients with pancreatitis from 31 families carried the N34S mutation (table 4). Of these, there was no N34S mutation in 36 subjects from 22 families with mutant PRSS1. Only one of 17 subjects from six families with wild-type PRSS1 (6%) carried the N34S mutation. Finally, there were four (6%) of 67 patients with ACP who carried the N34S mutation (p=0.235 v controls).
N34S mutation status and clinical outcome in patients with hereditary and idiopathic pancreatitis
The presence of the N34S mutation was not associated with early disease onset or any aspect of disease severity. Median (IQR) age at symptom onset in patients with HP was 12 (6.0–19.0) years and 12 (7.5–20.5) years for patients with ICP (log rank p value after Kaplan-Meier analysis; p=0.981). Comparisons of patients with and without the N34S mutation revealed no significant differences in terms of age of disease onset, including all patient groups together (log rank, p=0.279) or any of the groups separately (all p>0.05, data not shown).
Similarly, there were no significant differences between carriers and non-carriers of the N34S mutation in terms of age at onset of pancreatic exocrine insufficiency, age at onset of pancreatic endocrine insufficiency, duration of attacks, frequency of attacks, number of hospital admissions, surgery for pancreatic complications, or age at first surgery for pancreatic complications (all p>0.05, data not shown).
DISCUSSION
This study confirms an association between the SPINK1 N34S mutation and ICP.16,22 The prevalence of N34S was higher in patients with ICP (18%) than in normal individuals (2.5%) and much higher in f-ICP patients (86%) than in t-ICP patients (12.6%). Moreover, we identified N34S in unaffected individuals: in family members of patients with pancreatitis as well as in controls. The prevalence of N34S was not significantly elevated in HP patients with a causative PRSS1 gene mutation (1%).
The combined prevalence of N34S in our control populations is consistent with the prevalence of 1.58% in individuals from the USA21 and 1.5% from France20 but contrasts with the lower prevalence rate of 0.36% from the Berlin area.16 Thus the prevalence of N34S is much more geographically varied than originally perceived.16,20,21 The prevalence of N34S compares with a much lower prevalence of ICP (∼0.0066%27). These observations argue against the notion that N34S is causative of ICP in an autosomal dominant manner (since an extremely low penetrance would be required) and support the previous linkage study.21 Witt and colleagues16 reported on a unique pancreatitis pedigree with a SPINK1 mutation (M1T) disrupting the start codon that conceivably could have had a dominant pattern of inheritance. Thus much more extensive populations need to be studied with respect to SPINK1 mutations than heretofore has been possible16,20,21 which first requires important ethical issues to be addressed.28
The N34S mutation was not associated with early disease onset or any aspect of disease severity. In contrast, Pfützer and colleagues21 found that of 88 patients with wild-type PRSS1, 21/24 (88%) patients with SPINK1 mutations had developed symptoms by the age of 20 years compared with only 41/64 (64%) patients with wild-type SPINK1 (p<0.05). They found relatively more cases of ICP with SPINK1 mutations (40.4% v 18%) although the proportion of HP cases with wild-type PRSS1 and N34S were similar (9.1% v 11.1%, respectively). Moreover, we failed to identify any significant phenotypic or genotypic differences between patients with ICP and wild-type PRSS1 HP.
It seems logical to postulate that functional mutations of SPINK1 might predispose to pancreatitis given that ∼20% of trypsin activity is blocked by this inhibitor.19,29 Nevertheless, the only evidence that the N34S mutation may prevent the functional interaction between activated trypsin and SPINK1 to inhibit trypsin activity is based on the physiology of the porcine homologue and structural modelling of the human enzymes.21
The proposal that patients with chronic pancreatitis may suffer from an autosomal recessive disease due to mutant SPINK116 cannot be supported, as individuals who are heterozygous for N34S ought to be asymptomatic. Clearly, this is not the case, as shown by the high prevalence of affected individuals who were N34S heterozygotes in this and another study.21 The alternative proposal that the N34S mutation may interact with some other gene16 begs the question of which gene and how. The N34S gene cannot explain the ≥80% penetrance of mutant PRSS1 as the prevalence of N34S was only 1% in HP families with mutant PRSS1. Another possibility is the cystic fibrosis transmembrane conductance regulator (CFTR) gene, mutations of which have been associated with up to 37% of patients with ICP30–33 and up to 10% of patients with ACP.30 In Liverpool, the prevalence of major CFTR mutations is ∼5% and N34S ∼4%, so 0.2% of the population would be expected to have both N34S and a CFTR mutation and hence pancreatitis but this is not the actual case. Clearly, the CFTR gene is not a good candidate for the second gene although a particular CFTR mutation might be a possibility. Witt and colleagues34 recently reported a 5.8% prevalence of N34S in patients with ACP (similar to the 6% prevalence in this study) that was significant compared with their controls (0.8%) and further implicates N34S in the pathogenesis of pancreatitis.
From the present study we could not support the view that mutant SPINK1 is directly causative of pancreatitis,16,22 nor could we support the view that mutant SPINK1 is simply a disease modifier.21 Segregation of the N34S mutation in patients with pancreatitis is unexplained. The results from this study suggest an association between N34S and another gene in the pathogenesis of chronic pancreatitis. This may be the result of N34S increasing the penetrance of a gene that predisposes to pancreatitis or it could be that N34S is merely more prevalent in a population that for other reasons has a high incidence of pancreatitis. There is a developing consensus that the pathological role of N34S in pancreatitis is more complex than originally envisioned.35,36
This work was funded by grants from: the Northwest Cancer Research Fund, UK; MRC Gastroenterology and Pancreas Research Co-operative Grant; Solvay Health Care GmbH, Germany; the Augustus Newham Foundation; NIH, USA grant NIDDK-DK54709; USA Veterans Administration Merit Review grant; IZKF, Münster and the DFG, Germany.
Characteristics of the study population
Summary of N34S mutations identified
Summary of the distribution of the N34S mutation in affected individuals
Summary of the distribution of the N34S mutation in unaffected relatives of affected individuals
Contingency table comparing the incidence of N34S mutations between different study population groups
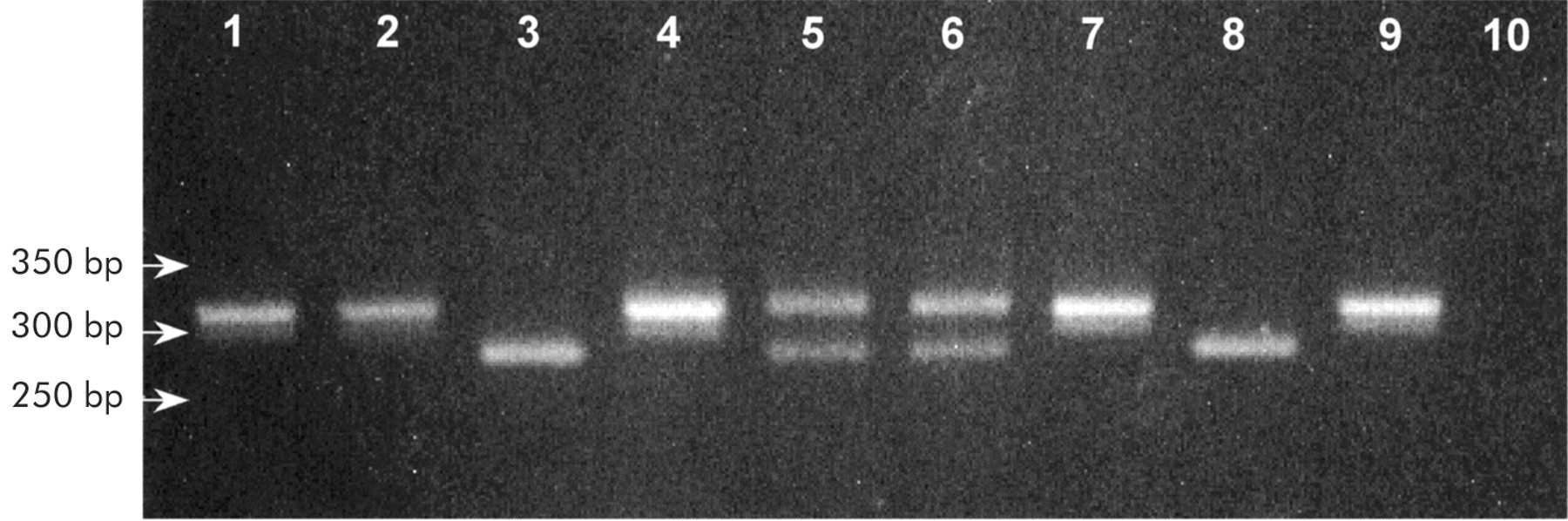
N34S mutation restriction fragment length polymorphism analysis. Gel electrophoresis (3% Nusieve) of PstI and BsrDI digestion products obtained from a wild-type individual, a heterozygote, and a homozygote. Lanes 1, 4, 7: undigested polymerase chain reaction product. Lane 2: wild-type individual—PstI. Lane 3: wild-type individual—BsrDI. Lane 5: heterozygous individual—PstI. Lane 6: heterozygous individual—BsrDI. Lane 8: homozygous individual—PstI. Lane 9: homozygous individual—BsrDI. Lane 10: negative control.

{kind=link}
{kind=link}
Family tree of a family with familial idiopathic chronic pancreatitis. Two (III:2 and III:3) of three affected patients and one (II:11) of nine unaffected individuals were carriers of N34S. Age at symptom onset for the two N34S positive patients was 17and 12 years, respectively. The unaffected individual with N34S (II:11) was their 64 year old mother. The third (III:4) affected individual, who was not tested for the N34S mutation, was diagnosed with pancreatitis at the age of 12 years. No confirmed cases of pancreatitis have been recorded in generations I or II. Two individuals on the paternal side of the family have been diagnosed with diabetes mellitus (II:6 was diagnosed at the age of 57 years and II:10 at 30 years). Neither of these two patients has complained of abdominal pain. The father of the affected individuals reported that his mother (I:1) suffered from abdominal pain; I:1 is now deceased and there was no known diagnosis of pancreatitis. On the maternal side, there was no report of any symptoms consistent with pancreatic disease.
Acknowledgments
The European Registry of Hereditary Pancreatic Diseases: The study coordinator, EUROPAC, Department of Clinical Genetics, Alder Hey Children's Hospital, Eaton Road, Liverpool, L12 2AP, UK. europac{at}liv.ac.uk; http://www.liv.ac.uk/surgery/europac.html.
Belgium: Professor J Deviere, Prof W van Steenbergen, Dr G Veereman-Wauters. Czech Republic: Dr J Martinek. Denmark: Dr T Havelund, Professor OB Schaffalitzky, Dr P Schmidt. France: Professor R Laugier. Germany: Dr W Boeck, Professor S Endres, Professor TM Gress, Professor MM Lerch, Dr C Niederau, Dr S Peters. Greece: Professor S Nousia-Arvanitakis. Hungary: Dr A Oláh, Dr V Ruszinko. Ireland: Dr D Barton, Professor B Drumm, Professor A Green, Dr C Imrie, Dr S Jaffrey, Dr F Murray, Dr O' Donnell, Dr Tanner, Dr D Vaughan. Italy: Professor Cavallini, Dr L Frulloni, Dr V Lucidi, Dr Salerno, Dr A Staiano, Professor G Uomo. Netherlands: Professor J Jansen. Norway: Professor Å Andren-Sandberg. Portugal: Dr Baldaia. Spain: Dr E Domunez-Munoz. Sweden: Professor I Ihse, Dr M Soller. Switzerland: Professor RW Ammann, Dr R Rossi, Dr K Truninger. UK: Dr A Adamson, Dr Ahliwalia, Mr RJL Anderson, Dr AM El Badri, Professor Benjamin, Dr CP Bennett, Mr D Berry, Professor J Booth, Dr E Boyd, Mr BJ Britton, Mr P Burgess, Dr S Cairns, Mr R Carter, Dr S Chambers, Mr R Charnley, Dr AT Cole, Professor D Colin-Jones, Dr J Collier, Dr M Cosgrove, Miss J Creighton, Mr WJ Crisp, Dr M Dalzell, Mr Davenport, Professor BR Davidson, Mr A Dennison, Dr D Dungar, Dr D Eccles, Dr D Fine, Dr NDC Finlayson, Dr CF French-Constant, Professor I Gilmore, Dr M Green, Mr R Hall, Dr Hatfield, Dr Z Hatton, Dr S Hodgson, Professor CW Imrie, Mr CD Johnson, Dr R Jones, Dr JY Kang, Professor A Kingsnorth, Mr T Leese, Dr DI Lewis-Jones, Dr K Lindley, Dr M Lombard, Ms M MacCullagh, Dr A Makin, Mr R McCloy, Dr PJ McKiernan, Dr A Mee, Dr V Murday, Dr S Murphy, Dr K Murray, Dr Muscroft, Ms M O'Donnell, Dr G Owen, Mr J Patterson, Dr S Prothero, Dr Pawinski, Dr GC Rastogi, Mr C Russell, Dr JD Sanderson, Mr NM Sharer, Dr J Shearman, Dr F Shek, Mr AS Sigurdsson, Dr R Simpson, Mr J Slavin, Dr H Smart, Professor P Sonksen, Mr Steinbrecker, Dr Sullivan, Mr M Super, Mr R Sutton, Dr JC Symons, Dr K Temple, Dr H Thomas, Mr J Thompson, Dr AJ Turnbull, Dr HJ Vadeyar, Dr M Veesey, Dr AC Wicks, Dr ML Wilkinson, Dr P Wilson.