Article Text

Abstract
Background and aims: Persistent inflammation observed in inflammatory bowel disease may be the consequence of an increased or aberrant immune response to normal gut constituents or an overall immune dysregulation and imbalance. Cytokines play an important role in immune regulation and interleukin 18 (IL-18) is one such cytokine that has emerged as being instrumental in driving CD4+ T cell responses towards a Th1-type. IL-18 can also directly mediate inflammation, moderate interleukin 1 activity, and can act on cell types other than T cells. It has been reported recently that IL-18 mRNA and protein are upregulated in gut tissue from IBD patients. The aim of this study was to understand more about the role of IL-18 in contributing to the pathology of IBD and to assess whether blocking IL-18 activity may be of therapeutic benefit as a treatment regimen for IBD.
Methods: Mice with dextran sulphate sodium (DSS) induced colitis were treated with recombinant IL-18 binding protein (IL-18bp.Fc), a soluble protein that blocks IL-18 bioactivity. Histopathological analysis was performed and RNA from the large intestine was analysed using the RNase protection assay and gene arrays.
Results: IL-18 RNA levels increased very early in the colon during DSS colitis. Treatment of mice with IL-18bp.Fc inhibited IBD associated weight loss and significantly inhibited the intestinal inflammation induced by DSS. IL-18bp.Fc treatment also attenuated mRNA upregulation of multiple proinflammatory cytokine genes, chemokine genes, and matrix metalloprotease genes in the large intestine that are commonly elevated during IBD.
Conclusions: IL-18bp treatment attenuated inflammation during DSS induced colitis in mice. Neutralising IL-18 activity may therefore be of benefit for ameliorating the inflammation associated with human intestinal diseases.
- animal model
- mucosa
- inflammation
- dextran sulphate sodium colitis
- interleukin 18
- CD, Crohn's disease
- UC, ulcerative colitis
- DSS, dextran sulphate sodium
- IBD, inflammatory bowel disease
- IL, interleukin
- IL-18bp, interleukin 18 binding protein
- IL-1ra, interleukin 1 receptor antagonist
- ICE, interleukin 1 converting enzyme
- IFN-γ, interferon γ
- PBS, phosphate buffered saline
- RPA, RNase protection assay
- MMP, matrix metalloprotease
- TIMP, tissue inhibitor of MMPs
- MLN, mesenteric lymph node
- TNF-α, tumour necrosis factor α
- HuIgG, human IgG
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Statistics from Altmetric.com
- CD, Crohn's disease
- UC, ulcerative colitis
- DSS, dextran sulphate sodium
- IBD, inflammatory bowel disease
- IL, interleukin
- IL-18bp, interleukin 18 binding protein
- IL-1ra, interleukin 1 receptor antagonist
- ICE, interleukin 1 converting enzyme
- IFN-γ, interferon γ
- PBS, phosphate buffered saline
- RPA, RNase protection assay
- MMP, matrix metalloprotease
- TIMP, tissue inhibitor of MMPs
- MLN, mesenteric lymph node
- TNF-α, tumour necrosis factor α
- HuIgG, human IgG
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Crohn's disease (CD) and ulcerative colitis (UC) are chronic inflammatory bowel diseases (IBD) characterised by chronic inflammation of the gastrointestinal tract.1 The recent establishment of animal models for IBD has provided evidence that immune dysregulation and altered cytokine secretion patterns may play a role in the pathology of IBD.2 Two cytokines that are thought to be important factors in the inflammatory cascade are interleukin (IL)-1 and IL-18.3 IL-1 has already been established as an important mediator of inflammation in multiple models of autoimmunity, including collagen induced arthritis and experimental autoimmune encephalomyelitis, as well as IBD.4 On the other hand, relatively little is known of the role of IL-18 in the inflammatory cascade in vivo, although recent reports describe increased IL-18 RNA and protein in tissues from IBD patients compared with tissues from normal patients.5,6 Furthermore, administration of IL-18 together with IL-12 has been shown to induce intestinal inflammation in mice.7
IL-18 was initially identified by its ability to stimulate interferon γ (IFN-γ) production in T cell lines and further characterisation has revealed IL-18 to be a primary mediator that biases responses towards the Th1-type in vivo.8–10 IL-18 can also act on other cell types, such as natural killer cells. IL-18 activity appears to be regulated at many different levels. Firstly, IL-18 is synthesised as a non-functional precursor, pro-IL-18. Protease activity mediated by interleukin 1 converting enzyme (ICE) is needed for conversion of pro-IL-18 to the mature functional protein.11–13 Mature IL-18 is then stored within the cell and is released on cell activation. The activity of IL-18 can also be regulated after release from the cell. A naturally occurring protein, interleukin 18 binding protein (IL-18bp), that is normally present in serum can bind IL-18 and block bioavailability and subsequent function.14 Tight regulation of mature IL-18 production, as well as regulation of its availability in serum, suggests that it may be an important inflammatory mediator.
The complex interplay between different cytokines can provide another tier of immune regulation, either by providing synergy or counterregulation. For IL-18, it is clear that more profound Th1 responses can be elicited when there is synergy with IL-12. Production of IFN-γ by T cells is amplified dramatically by stimulation with IL-12 and IL-18 together compared with either cytokine alone.15 IL-18 has also been shown to be symbiotically involved with IL-1 regulation. IL-18 and IL-1 are thought to regulate each other by both stimulating the production of functional ICE, which in turn regulates the downstream production of mature IL-1 and IL-18.12,16,17 Thus it seems that IL-18 plays an important role in the inflammatory cascade by regulating IL-1, in addition to being an important factor in the induction of Th1 responses.
In the current study, we were interested in determining whether initiation of intestinal pathology associated with IBD might in part be mediated by IL-18. Although there are many mouse models of IBD, not all appear to involve IL-18. Firstly, we assessed IL-18 RNA levels in the intestine in a number of different mouse IBD models. We consistently observed increased IL-18 RNA levels in the large intestine of mice administered dextran sulphate sodium (DSS) to induce acute colitis. We therefore chose to pursue our investigation using the acute DSS induced model of IBD in C57BL/6 mice. We tested the hypothesis that IL-18 may be an important mediator of inflammation in this IBD model by treating mice with a recombinant form of IL-18bp to block IL-18 activity during the disease process. We assessed a number of disease parameters, including weight loss and histopathology, and analysed RNA from the large intestine for cytokine gene expression. We used array technology to assess local regulation of chemokine, chemokine receptor, and matrix metalloprotease (MMP) genes during experimentally induced IBD, and determined which of these genes could be counterregulated by IL-18bp treatment during colitis. We showed that blocking IL-18 in vivo is an effective method of attenuating experimentally induced colitis. These data suggest that IL-18 is an important mediator of the initial inflammatory events in the intestine.
MATERIALS AND METHODS
Mice
Female C57BL/6 mice were purchased from Taconic Laboratories (Germantown, New York, USA). All animals were fed standard laboratory chow, given water ad libitum, and maintained in a specific pathogen free facility at Immunex Corporation, in accordance with approved ethical guidelines. All mice were 8–10 weeks of age at initiation of the experiments and were age matched within experiments.
DSS induced colitis
Colitis was induced in mice by administering 2% (wt:vol) dextran sodium sulphate (DSS, molecular weight 40 000; ICN Biochemicals Inc., California, USA) from day 0 to day 7 in the drinking water ad libitum followed by return to normal water. Mice were weighed every day beginning on day 0 and weight changes were recorded until day 13. Per cent weight change for each individual mouse was calculated as follows: % weight change=((weight at specific day (day 0−day 13)−weight on day 0)/weight on day 0)×100.
Production and purification of IL-18bp.Fc
The huIL-18bp was cloned from an in house human dendritic cell library using primers described previously.14 Primers used for amplification were: forward primer 5`CCTAGCAAGTCGACCAGGCTCACCAGCTCCTGACGC 3`; reverse primer 5` GCTACTAGATCTACCCTGCTGCTGTGGACTGCTGTGGC 3`. The IL-18bp.Fc fusion molecule was generated by joining the entire IL-18bp cDNA to the CH2 and CH3 domains of human IgG1 (HuIgG1) in an expression plasmid, as previously described.18 Briefly, CV-1/EBNA cells were transiently transfected with the pDC412/huIL-18bp.Fc expression plasmid. Supernatant was collected from the cells one week later and protein purified by passing over a protein A resin column (POROS20 from Perseotive Biosystems). The protein was eluted with 50 mM citrate, pH 3.0, and dialysed using 7000 MWCO dialysis tubing. All batches of recombinant IL-18bp used in these experiments were subjected to N terminal sequencing, were devoid of contaminating protein, and were deemed endotoxin free (< 10 EU/mg). In vitro bioassays were used to confirm that the recombinant IL-18bp.Fc was efficient in blocking the bioactivity of both human and murine IL-18 (data not shown).
In vivo treatment protocol and reagents
Groups of mice were treated intraperitoneally with huIL-18bp.Fc (60 or 600 μg) or HuIgG (60 or 600 μg) as a control protein (Sigma-Aldrich Co., St. Louis, Missouri, USA), each in a total volume of 200 μl per injection. Control HuIgG was ineffective at modulating IL-18 activity. All reagents were resuspended in endotoxin free phosphate buffered saline (PBS) and tested for endotoxin before use (<100 pg/injection). Mice were injected intraperitoneally from day 0 to day 7 of the experiment with the reagents specified. Statistical analysis was performed with the Student's t test using InStat software (Graphpad Software, San Diego, California, USA).
Tissue harvest
Mice were sacrificed by cervical dislocation and tissues removed and processed for RNA preparation, histopathology, and cytokine analysis. For the initial analysis of the kinetics of gene regulation during DSS induced colitis, tissues were collected on days 0, 2, 4, 6, and 8 following the start of DSS treatment. For the in vivo experiments using IL-18bp.Fc treatment, tissues were collected on day 8 of the DSS treatment regimen.
Histopathological analysis
Tissue was fixed in formalin, paraffin embedded, sectioned, and then stained with haematoxylin and eosin. Studies were analysed by a pathologist (SK) using a disease scoring system. Histopathological changes were individually scored as 0 (no findings), 1 (minimal), 2 (mild), 3 (moderate), or 4 (severe) for each of the parameters described in table 1. All slides were blinded for scoring. Each mouse was scored individually for each of the parameters described and the scores averaged (n=4–6 mice/group).
Pathology scores: interleukin 18 binding protein (IL-18bp.Fc) treatment inhibits dextran sulphate sodium (DSS) induced colitis
Preparation of RNA
Large intestines were harvested from groups of 2–6 mice, pooled, and homogenised in guanidium isothiocyanate buffer (4.5 M guanidium isothiocyanate, 50 mM sodium citrate, 0.5% (wt/vol) sodium sarcosyl containing 2% 2-mercaptoethanol). RNA was extracted from cell lysates by standard methods using cesium chloride (Maniatis et al, Cold Spring Harbour). Concentration of RNA was determined by absorbance at 260 nm and RNA quality assayed by running 1 μg of RNA on a 0.5% TBE gel.
RNase protection assay (RPA)
Cytokine mRNA levels were measured using Riboquant Multiprobe RNase Protection Assay kits mCK-2b and mCK-3b (Pharmingen, San Diego, California, USA). The RPA was carried out according to the manufacturer's instructions using 20 μg of RNA from each set of tissue samples. Products were visualised on a 6% urea gel and quantitated using phosphorimaging analysis and software (Molecular Dynamics, Sunnyvale, California, USA). The relative expression of each cytokine was normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for each individual sample.
Gene array analysis
Expression profiles were established by hybridisation of labelled RNAs to Mu11K chip sets (Affymetrix, Santa Clara, California, USA), consisting of approximately 11 000 mouse genes and EST clusters from Unigene (Build 4) and 800 EST clusters from TIGR (1.0 Beta). Total RNA (5 μg) from individual samples from the large intestine were labelled according to standard protocols (Affymetrix) and hybridised to duplicate arrays in parallel. Hybridised chips were processed as recommended and scanned images analysed using Genechip software (Affymetrix). Data were migrated to an Oracle database and mined using Spotfire 4.0 (Spotfire Inc., Cambridge, Massachusetts, USA). All data are expressed as a fold change in mRNA levels relative to the no DSS treatment group. The fold changes presented were calculated from averaging the four comparative analyses generated by Genechip from the duplicates for a particular pair of RNAs.
Western analysis
Western analysis was performed using standard protocols. Briefly, large intestines were lysed in HNTG lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM MgCl2, 10% glycerol, 1% Triton X-100, proteinase inhibitor cocktail). Equivalent amounts of total protein were then incubated with 30 μg anti-IL-18 antibody C-18 conjugated agarose beads (C-18 AC; Santa Cruz Biotechnology Inc, Santa Cruz, California, USA). The resulting immunoprecipitated samples were subject to western analysis by separating on 14% Tris-glycine gels prior to transfer onto PVDF membranes (Invitrogen, Carlsbad, California, USA) using standard protocols. Tris buffered saline substituted with 1% Tween-20 and 5% NFDM was used to block non-specific binding to the membrane, prior to incubation with a rabbit anti-IL-18 polyclonal mAb (H-173; Santa Cruz Biotechnology Inc) at 1:2500 dilution. The membrane was washed and incubated with anti-rabbit peroxidase at 1:10 000 dilution (Jackson Immunochemicals, Westgrove, Pennsylvania, USA), washed and developed using ECL-Plus peroxidase developing solution (Amersham Pharmacia Biotech, Buckinghamshire, UK). As a positive control, recombinant murine IL-18 (MBL, Nagoya, Japan) was used at 5 ng by itself or was spiked into intestine lysates to serve as a positive control for immunoprecipitation.
Cell staining
Mesenteric lymph node (MLN) cells were isolated from individual mice from the different treatment groups and viable cell counts obtained by trypan blue dye exclusion. For analysis of cell types within the MLN population, 1×106 cells were costained using fluorescein conjugated anti-CD4 and phycoerythrin conjugated anti-CD8 mAbs using standard cell staining protocols, as described previously.19 All antibodies were obtained from BD Pharmingen (San Diego, California, USA). Cell analysis was done using a Becton Dickinson FACScan (Becton Dickinson, San Jose, California, USA). A minimum of 104 events per sample were collected for analysis.
Cytokine ELISA analysis
For cytokine protein analysis, 3×106 cells were cultured in 24 well plates at 37°C in 5% CO2 for 48 hours. Plates were preincubated with either PBS or anti-CD3 (clone 500A2, 10 μg/ml) 4°C, overnight. Supernatants from the cell cultures were harvested and ELISAs for mouse IL-4, IL-10, and IFN-γ performed using standard protocols, as specified by the manufacturer. All purified coating and biotinylated capture antibodies were obtained from Pharmingen (San Diego, California, USA). The streptavidin-horseradish peroxidase conjugate was obtained from Zymed (San Francisco, California, USA). The horseradish peroxidase substrate solutions were obtained from KPL (Gaithesburg, Maryland, USA). The ELISA plates were read using a Molecular Devices Thermomax plate reader and analysed using the Deltasoft software analysis system. ELISA was performed on samples obtained from individual mice and the data shown are the average for the group (n=4–6 mice/group).
RESULTS
IL-18 RNA is upregulated in the large intestine of mice during the early stages of DSS colitis
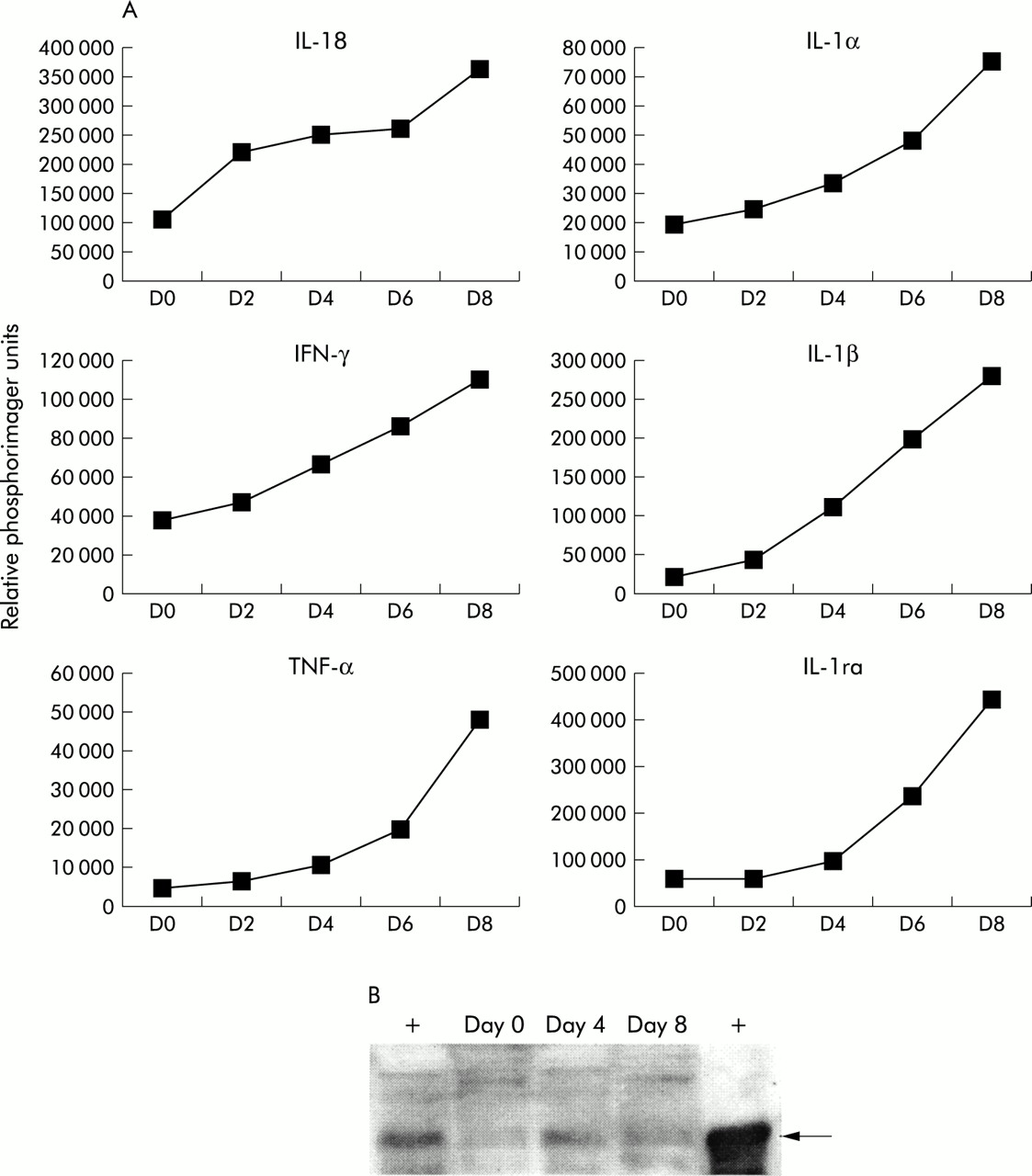
DSS is commonly used in animal models to induce inflammation in the gastrointestinal tract.20,21 Susceptibility to DSS varies between mouse strains.20,21 C57BL/6 mice are very sensitive to DSS and severe intestinal inflammation can be induced using doses of DSS as low as 2–3% (w/v).20,21 To identify the temporal regulation of genes during induction of DSS induced colitis, we administered 2% DSS to C57BL/6 mice from day 0 to day 7. DSS was then replaced with normal drinking water. Groups of mice were sacrificed on days 0, 2, 4, 6, and 8. RNA from the large intestine was isolated and RPA analysis performed. We observed an increase in IL-18 RNA in C57BL/6 mice beginning on day 2 after the onset of DSS administration (fig 1A), indicating that IL-18 may play an important role in the early inductive stages of DSS induced colitis. We verified the presence of increased IL-18 protein during DSS induced colitis by western analysis. As shown in fig 1B, there was an increase in mature IL-18 protein on days 4 and 8 of DSS treatment compared with day 0. IL-1α, IL-1β, interleukin 1 receptor antagonist (IL-1ra), tumour necrosis factor α (TNF-α), and IFN-γ mRNA were also upregulated concordant with induction of DSS induced colitis but gross upregulation was not observed until day 4 after the onset of DSS administration. The pattern of regulation of each of these genes during DSS colitis is shown in fig 1A. These results show that IL-18 was upregulated in the early phase of DSS colitis and preceded upregulation of IL-1α, IL-1β, TNF-α, and IFN-γ. Similar gene regulation profiles were seen in BALB/c mice treated with 5% DSS (data not shown).

Interleukin (IL)-18 mRNA was upregulated in the large intestine in the early stages of dextran sulphate sodium (DSS) induced colitis. (A) C57BL/6 mice were treated with 2% DSS in water from day 0 to day 7. Mice were sacrificed on days 0, 2, 4, 6, and 8 (3 mice/time point). Large intestine RNA was isolated and the RNase protection assay was performed using the MCK2b and MCK3b probe sets from Pharmingen. Data are shown as relative phosphorimager units normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. Data are representative of two separate experiments. IFN-γ, interferon γ; TNF-α, tumour necrosis factor α; IL-1ra, interleukin 1 receptor antagonist. (B) C57BL/6 mice were given 2% DSS in water from day 0 to day 7. Mice were sacrificed on days 0, 4, and 8 (2 mice/time point) and the amount of mature IL-18 protein in the large intestine was assessed by western analysis. Recombinant murine IL-18 was used as a positive control. Left lane, immunoprecipitation from IL-18 spiked intestinal lysate; right lane, recombinant IL-18.
Blocking IL-18 in vivo with IL-18bp.Fc inhibits weight loss during DSS induced colitis
Administration of DSS to mice induced transient weight loss associated with development of colitis, followed by recovery (fig 2). As IL-18 RNA was found to be upregulated in the large intestine during the early stages of DSS induced colitis, we wished to examine the possibility of inhibiting weight loss associated with intestinal inflammation by blocking IL-18 function. IL-18bp has been shown to bind and inhibit IL-18 function in vitro.14,18,22 In order to block IL-18 function in vivo, IL-18bp.Fc was administered daily by intraperitoneal injection beginning on day 0, and continuing until day 7. We used two different doses of IL-18bp.Fc (600 μg/injection and 60 μg/injection). As shown in fig 2, the high dose of 600 μg/injection of IL-18bp.Fc inhibited weight loss significantly (by up to 85% inhibition on day 9; p<0.001) compared with HuIgG injected control mice given DSS. Experiments with the lower dose of IL-18bp.Fc (60 μg/injection) had a less significant but reproducibly observable effect on inhibiting DSS associated weight loss (approximately 30–50% inhibition). IL-18bp.Fc treatment had no effect on the weights of control mice not treated with DSS. These data reveal that blocking IL-18 function in vivo can inhibit weight loss seen during DSS induced colitis, one parameter used to measure intestinal inflammation in animals.

Interleukin 18 binding protein (IL-18bp.Fc) treatment inhibited weight loss associated with dextran sulphate sodium (DSS) induced colitis. C57BL/6 mice were treated with 2% DSS in water from day 0 to day 7. Mice were injected intraperitoneally with IL-18bp.Fc (60 or 600 μg/day) or control human IgG (HuIgG 600 μg/day) from day 0 to day 7. The weight of individual mice was followed daily. The data shown are the average per cent weight change for each group (n=6 mice/group) and are representative of three separate experiments.
Blocking IL-18 function in vivo with IL-18bp.Fc inhibits pathological inflammation in the large intestine of mice given DSS
One of the major hallmarks of DSS induced colitis is the profound inflammation in the colon of animals, characterised by crypt destruction, mucosal ulceration, erosions, and infiltration of lymphocytes into the mucosal tissue.23 In agreement with previous studies, we observed that the entire colon was affected, with loss of crypt regions evident by day 8 of DSS treatment (table 1). Colitis was associated with massive inflammatory cell infiltration composed of lymphocytes and neutrophils, giving rise to severe ulceration of the mucosa (fig 3B, table 1). Colons from DSS treated mice typically contained 3–5 ulcers and the average total lesion score for the group was 19.3 (table 1). In contrast, histological analysis of the colon revealed that mice treated with IL-18bp.Fc during DSS colitis had significantly decreased inflammation and ulceration (p<0.001) (fig 3C, table 1). Mice treated with IL-18bp.Fc showed a marked decrease in the severity of the lesions in the colon, and number of ulcers ranged from 0 to 2 for each mouse, and the average total lesion score was approximately half that of the HuIgG control treated DSS group (table 1). Furthermore, animals treated with IL-18bp.Fc exhibited a significant reduction in infiltration by lymphocytes and neutrophils in the lamina propria (p<0.05 and p<0.01, respectively). These data clearly demonstrate that treating mice with IL-18bp.Fc attenuates not only weight loss but also the inflammatory changes in the colon associated with DSS induced colitis.

Interleukin 18 binding protein (IL-18bp.Fc) treatment inhibited inflammation associated with dextran sulphate sodium (DSS) induced colitis in the large intestine. Large intestines were removed from individual mice on day 8, fixed in formalin, paraffin embedded, sectioned and stained with haematoxylin and eosin. Representative sections of mouse large intestine from C57BL/6 mice are shown (100× original magnification). HuIgG, human IgG.
IL-18bp.Fc treatment during DSS colitis prevents upregulation of mRNA for IL-1α, IL-1β, TNF-α, IFN-γ, IL-1ra, but does not affect IL-18 mRNA
To further analyse the effects of blocking IL-18 during IBD, and to characterise more fully the regulation of genes during the process of DSS colitis, RNA was isolated from the large intestine of animals from the different treatment groups on day 8. Cytokine gene regulation was assayed using RPA. A representative RPA gel is shown in fig 4A. As expected, DSS colitic animals treated with control HuIgG show increased levels of IFN-γ, TNF-α, and increased levels of the IL-1 family proteins IL-1α, IL-1β, and IL-1ra (fig 4B). Treatment with IL-18bp.Fc during DSS treatment blocked upregulation of these genes. These data provide further evidence of a role for IL-18 in contributing to the pathology of DSS induced IBD. IL-18 RNA itself was however not regulated by IL-18bp.Fc treatment, indicating that transcription of IL-18 was not affected in IL-18bp.Fc treated mice. This suggests that IL-18bp does not regulate IL-18 by a transcriptional feedback mechanism.

Interleukin 18 binding protein (IL-18bp.Fc) treatment attenuated induction of proinflammatory cytokine mRNA during dextran sulphate sodium (DSS) induced colitis. RNA was isolated from the large intestine of C57BL/6 mice on day 8 from each of the treatment groups (n=6 mice/group). The RNase protection assay was performed using the MCK-2b and MCK-3b probe sets. A representative section of an RPA (MCK-2b probe set) is shown in (A). RNA levels for genes were quantitated using a phosphorimager. HuIgG, human IgG. The data shown in (B) were obtained from the groups receiving 600 μg IL-18bp.Fc or HuIgG. Data are expressed as relative phosphorimager units, normalised to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels, and are representative of two separate experiments. IFN-γ, interferon γ; TNF-α, tumour necrosis factor α; IL-1ra, interleukin 1 receptor antagonist.
In vivo treatment with IL-18bp.Fc prevents the increased cellularity and increased cytokine secretion in the MLN that is associated with IBD
One of the major characteristics of inflammation in the gastrointestinal tract is that the cellularity of the MLN, the major lymph node draining from the gut, increases compared with non-colitic mice (fig 5A). This change in cellularity is due to infiltrating mononuclear cells such as T cells and macrophages (data not shown). Analysis of T cell populations and numbers in the MLN revealed an increase in the relative proportion and absolute number of CD8+ T cells compared with CD4+ T cells (data not shown). IL-18bp.Fc inhibited the increased cellularity seen in the MLN of mice with DSS colitis (fig 5A) and analysis of the T cell populations and CD4+/CD8+ T cell ratios showed that the numbers and ratios of T cells in the MLN from IL-18bp.Fc treated mice were similar to non-DSS treated groups (data not shown).
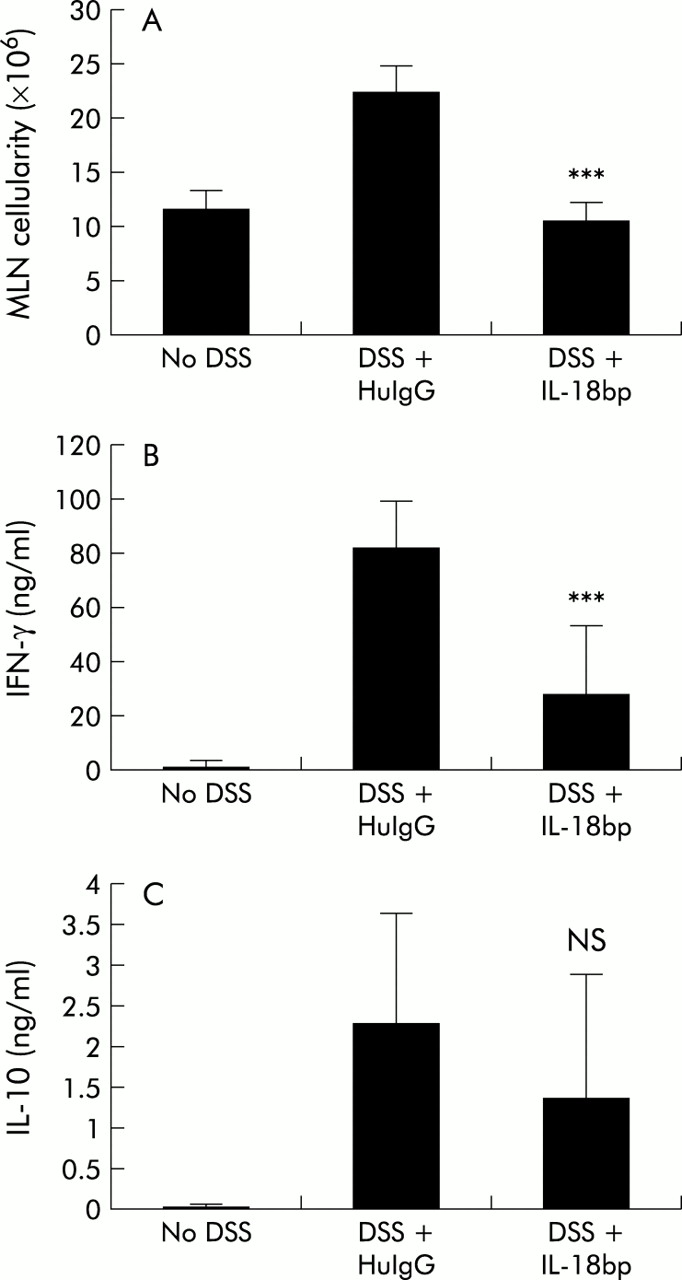
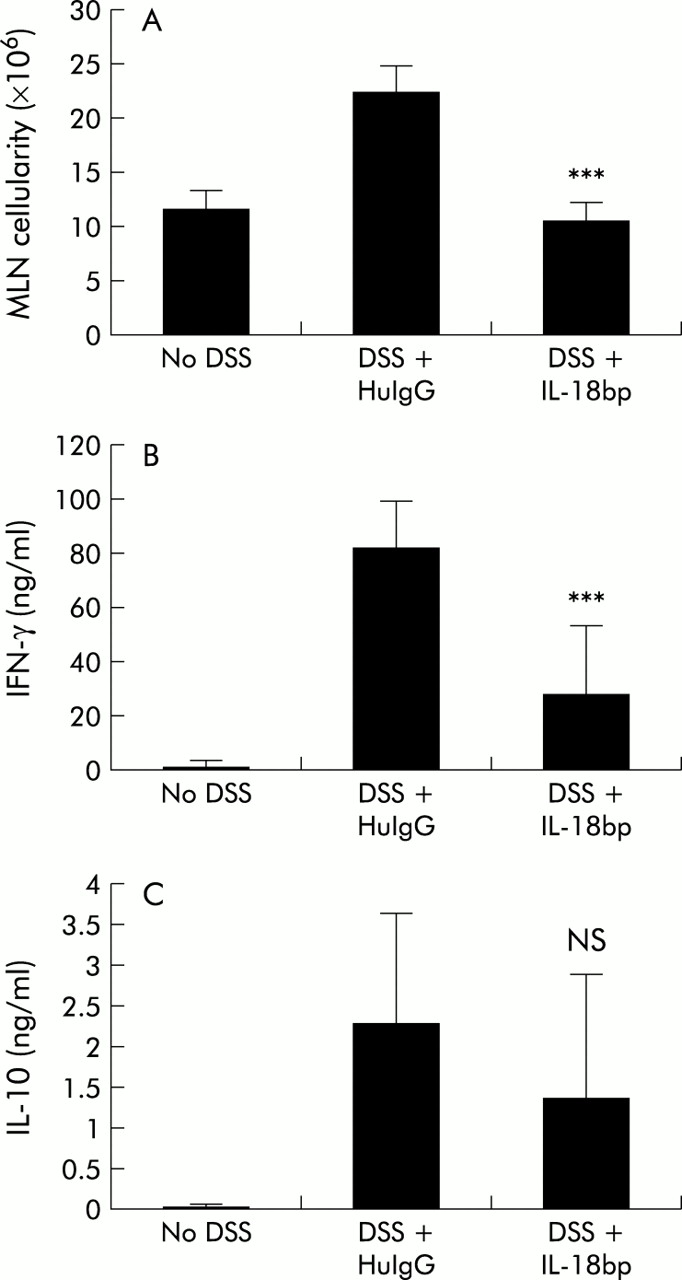
Interleukin 18 binding protein (IL-18bp.Fc) treatment inhibited the increased cellularity and increased cytokine production in the mesenteric lymph node (MLN) that is associated with dextran sulphate sodium (DSS) induced colitis in C57BL/6 mice. (A) Individual MLNs were isolated from mice from the various treatment groups. Viable cell count was obtained by trypan blue exclusion. The data shown are the average number of MLN cells/mouse (n=4 mice/group). HuIgG, human IgG. (B, C) MLN cells were cultured on anti-CD3 coated plates. Supernatants collected after 48 hours were assayed by ELlSA for interferon γ (IFN-γ) (B) and IL-10 (C). Similar data were obtained in two separate experiments.
Because cytokines have been shown to play an important role in multiple models of colitis and as we observed changes in cytokine mRNA profiles in the intestine, we were interested in determining whether cytokine protein profiles of cells draining from the gut would be modulated by IL-18bp treatment. We analysed MLN cells from the various groups of animals on day 8 for in vitro secretion of IL-4, IL-10, and IFN-γ. MLN cells were cultured on either PBS or anti-CD3 coated plates for 48 hours and the culture supernatants analysed by ELISA for cytokines. As shown in fig 5B and C, MLN taken from mice with DSS colitis showed increased levels of both IFN-γ and IL-10 protein production following anti-CD3 treatment. In contrast, MLN from IL-18bp.Fc treated DSS colitic animals did not show the same increase in levels of IFN-γ and IL-10 protein. Although protein levels were not identical to control non-DSS levels, the decrease for IFN-γ was significant (p=0.003). IL-4 was below detectable levels for all samples in these experiments (data not shown). These results show that treatment with IL-18bp inhibits the process of inflammation during DSS induced colitis, presumably by attenuating the trafficking of T cells into the MLN and thus attenuating the increased cytokine secretion in the gut associated lymphoid tissues.
Analysis of chemokine/chemokine receptor and MMP gene regulation in IL-18bp.Fc treated mice during DSS colitis
To further characterise an expanded set of genes, RNA from the large intestine of the animals from the various treatment groups (day 8) was used in array analysis using Affymetrix chips. Approximately 300 genes showed greater than threefold regulation after DSS treatment and counterregulation following IL-18bp.Fc treatment (data not shown).
Focusing on chemokine and chemokine receptor gene regulation, we observed increases in MIP1α, MIP1β, MIP2, RANTES, CCR2, and CCR5 during DSS colitis (fig 6A). As shown in fig 6A, treatment with IL-18bp.Fc attenuated upregulation of these genes indicating that IL-18bp treatment may act upstream and be able to block the initiation of the chemokine inflammatory cascade associated with colitis. We also saw upregulation of ENA-78 (24×) and MIG (20×) in the large intestine of DSS colitic mice. IL-18bp.Fc treatment downregulated expression of these genes (data not shown). These data are consistent with our histopathological analysis shown in table 1 that indicates less recruitment of cells to the mucosa in IL-18bp treated mice.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Interleukin 18 binding protein (IL-18bp.Fc) treatment inhibited chemokine, chemokine receptor, and matrix metalloprotease (MMP) mRNA upregulation that is associated with dextran sulphate sodium (DSS) induced colitis in the large intestine of C57BL/6 mice. RNA from the large intestine on day 8 of treatment (n= 6 mice/group) was used to perform cDNA array analysis using Affymetrix chips. Data are expressed as the relative fold change in mRNA levels in the sample group relative to the no DSS treatment group. (A) Chemokine (MIP1α, MIP1β, MIP2, and RANTES ) and chemokine receptor (CCR2, CCR5) gene regulation. HuIgG, human IgG. (B) MMP and tissue inhibitor of matrix metalloprotease (TIMP) gene regulation.
With regard to tissue repair and remodelling mechanisms during inflammation, a number of investigators have documented an increase in expression of a number of MMPs in human IBD, including stromelysins (MMP-3 and 10), gelatinase B (MMP-9), collagenases (MMP-1, 8, and 13), and type IV collagen, as well as increases in tissue inhibitor of metalloproteinase 1 (TIMP-1).24–28 Here we reported an increase in RNA levels for MMP-3, 7, 9, 10, 13, and TIMP-1 in the large intestine of mice treated with DSS. As shown in fig 6B, treatment with IL-18bp.Fc decreased mRNA levels for these MMPs and TIMP-1, to levels seen in control tissue, again suggesting that blocking IL-18 attenuates the damage incurred during the initiating inflammatory stages of IBD.
DISCUSSION
A number of mouse models have been established to try to understand more about IBD. These models have revealed an important role for both immune dysregulation and altered cytokine patterns.2,29 Multiple cytokines, including IFN-γ, TNF-α, and IL-1, have been shown to be critical mediators of pathology during inflammatory processes. IL-18 is a relatively new cytokine that has evolved as an important mediator of Th1 responses but it should also be noted that IL-18 has an additional role as an important regulator of inflammation that can act on other cell types and regulate IL-1 activity. Recently, IL-18 RNA and protein were reported to be upregulated in the tissues of IBD patients but not in control patients, suggesting a role for this cytokine during the inflammatory process in IBD.5,6
In an attempt to understand more about the role of IL-18 in IBD, we used the mouse model of DSS induced colitis in C57BL/6 mice. We chose this model because C57BL/6 mice are sensitive to DSS and we observed that IL-18 RNA and protein are upregulated in the large intestine during the very early stages of IBD before mRNA upregulation of other proinflammatory cytokines such as IL-1α, IL-1β, TNF-α, and IFN-γ. Recently, two other groups have also reported increased IL-18 RNA levels in mouse models of IBD.30,31 Collectively, these data would suggest that IL-18 may be playing a significant role in the early inductive stages of the inflammatory pathway that leads to IBD.
To assess the extent of the role of IL-18 in vivo in IBD, we used IL-18bp.Fc to block IL-18 function in the DSS model of colitis. Treatment with IL-18bp.Fc significantly attenuated DSS induced colitis in C57BL/6 mice. IL-18bp.Fc treatment inhibited weight loss associated with DSS induced colitis, and histopathological analysis showed that cellular infiltration, ulceration, crypt destruction, and oedema seen in the large intestine during DSS induced colitis was also reduced by IL-18bp.Fc treatment. Although there was still some evidence of inflammation in the IL-18bp.Fc treated animals, IL-18bp.Fc treatment decreased the pathology inflammation score by 50%. Gene regulation analysis using a combination of RPA analysis and Affymetrix array analysis also showed that IL-18bp.Fc treatment inhibited upregulation of many of the inflammatory genes seen during colitis. Cellular infiltration/activation in the MLN associated with IBD was also blocked by IL-18bp.Fc treatment as demonstrated by decreased numbers of infiltrating T cells and decreased levels of IFN-γ production. By administering IL-18bp.Fc, it appears that we were effectively able to attenuate the initiation of the inflammatory cascade.
The data presented above support the fact that IL-18 plays a significant role in the primary stages of inflammation in DSS colitis. As IL-18 and IL-1 can regulate each other, one potential pathway for IL-18 mediating its effects is via IL-1 induction. IL-1 has also been reported to be a potent mediator of inflammation in multiple disease models.4,32 Although IL-1 protein and RNA are upregulated in tissue samples from CD and UC patients,33 there are mixed reports in animal models of colitis regarding whether it is possible to attenuate IBD by blocking IL-1.4,34–36 We compared the kinetics of upregulation of IL-18 and IL-1 RNA in the large intestine of DSS treated animals, and observed that IL-18 RNA was elevated by day 2, before upregulation of IL-1α and IL-1β RNA which was not upregulated until day 4. Although IL-1α and IL-1β were upregulated considerably by DSS treatment, blocking IL-18 with IL-18bp.Fc treatment prevented this upregulation. IL-18 therefore could be acting at an earlier stage and upstream of IL-1 in the DSS model, and by inhibiting IL-18 function it appears possible to block initiation of the IL-1 dependent inflammatory cascade.
IL-18 is a potent amplifier of IFN-γ synthesis by T and natural killer cells.37 We have provided both RNA and protein evidence for increased IFN-γ levels in the gut and gut associated lymphoid organs. This increase in IFN-γ mRNA and protein levels in DSS treated mice was inhibited in IL-18bp.Fc treated animals. The specific role for IFN-γ or IFN-γ regulated genes in the pathology of DSS induced IBD is not known. Although TNBS induced colitis can be seen in IFN-γR1−/− mice,38 IL-12 induced IFN-γ increases intestinal inflammation in the acute DSS colitis model.39 IFN-γ regulated genes may regulate other inflammatory cascade proteins. For example, IFN-γ induced nitric oxide has been shown to have a role as a toxic effector molecule in a chronic DSS colitis model and blocking IFN-γ or nitric oxide synthase significantly inhibits chronic colitis in mice.40,41 In our array analysis, mRNA for nitric oxide synthase was upregulated during DSS colitis and was counterregulated after IL-18bp.Fc treatment (data not shown). It is therefore possible that nitric oxide is a mediator of inflammation in the DSS colitis model.
In recent years there has been a focus on the role of chemokines, chemokine receptors,42 and MMPs43 in inflammation such as IBD. Increased expression of MCP-1 and MCP-3 has been found in colonic tissue from patients with IBD,44,45 chemokines which bind to the chemokine receptor CCR2. Increased expression of MIP1α, MIP1β, and RANTES has also been reported in the intestinal mucosa during IBD in humans,44,46,47 chemokines which bind to the chemokine receptor CCR5. Of note, it has recently been shown that mice deficient in CCR2 or CCR5 are protected from DSS induced intestinal lesions and mucosal ulcerations.48 In our current studies presented here, we observed increased expression of MIP1α, MIP1β, RANTES, ENA-78, MIG, CCR2, and CCR5 in the large intestine during DSS colitis. Upregulation of these chemokines and chemokine receptors was inhibited in mice treated with IL-18bp-Fc during colitis.
It has also been reported that increased levels of MMPs can be found in inflamed intestinal tissue. In particular, increased levels of stromelysin-1 (MMP-3), collagenases (MMP-1), and gelatinase B (MMP-9) have been reported in tissue samples from human IBD patients.24,26–28,49 In our mouse DSS colitis model, we observed increased expression of many different MMPs, including MMP-3, 7, 9, 10, and 13 concordant with intestinal inflammation. Upregulation of these MMPs was inhibited in mice treated with IL-18bp.Fc during colitis.
In summary, we have clearly demonstrated that blocking IL-18 function in vivo using IL-18bp.Fc is an effective method for attenuating intestinal inflammation induced by DSS in mice. The data presented above indicate a role for the IL-18 pathway in the initiation of intestinal damage associated with IBD. This information can be used to develop new therapeutic approaches for treating human IBD.
Acknowledgments
The authors would like to thank Dr Sharon Wong-Madden and Melissa Petersen for the production and purification of IL-18bp.Fc protein. We thank Bill Brady for assistance with the Affymetrix array experiments. We thank the histology group for their help in processing the slides. We thank Dirk Anderson for making the inhouse dendritic cell library from which IL-18bp was cloned. We also appreciate the helpful discussions provided by Dr John E Sims. We thank Drs John E Sims, David Cosman, and Phil Morrissey for critical review of the manuscript. We also thank Laurel J Nelson for editorial suggestions.
REFERENCES
Footnotes
-
Conflict of interest: All authors are employees of Immunex Corporation, Seattle, Washington, USA