Article Text

Abstract
Background and aims: C-myc over expression is implicated in malignancy although to date this has not been studied in Barrett’s metaplasia. We sought to determine c-myc expression in the malignant progression of Barrett’s metaplasia and whether it may be induced by bile acids seen in gastro-oesophageal refluxate.
Methods: C-myc protein and mRNA levels were assessed in 20 Barrett’s metaplasia and 20 oesophageal adenocarcinoma samples by western blotting and real time polymerase chain reaction. Levels of c-myc and proliferation were also assessed in cell lines OE21, OE33, SW-480, and TE-7 stimulated with pulses or continuous exposure to the bile acids deoxycholic acid and chenodeoxycholic acid.
Results: C-myc protein was upregulated in 50% of Barrett’s metaplasia and 90% of oesophageal adenocarcinoma samples compared with squamous, gastric, and duodenal controls. C-myc immunolocalisation in Barrett’s metaplasia revealed discrete nuclear localisation, becoming more diffuse with progression from low to high grade dysplasia to adenocarcinoma. Both continual and pulsed bile acid induced c-myc at pH 4, with no effect at pH 7 or with acidified media alone. Pulsed bile acid treatment induced proliferation (p<0.05); in contrast, continuous exposure led to suppression of proliferation (p<0.05).
Conclusions: We have shown upregulation of c-myc with malignant progression of Barrett’s metaplasia and suggest that acidified bile may be a novel agent responsible for induction of this oncogene.
- c-myc
- Barrett’s metaplasia
- bile acid
- GORD, gastro-oesophageal reflux disease
- DCA, deoxycholic acid
- CDCA, chenodeoxycholic acid
- PCR, polymerase chain reaction
- Ct, cycle threshold
- ESA, epithelial cell specific antigen
Statistics from Altmetric.com
- GORD, gastro-oesophageal reflux disease
- DCA, deoxycholic acid
- CDCA, chenodeoxycholic acid
- PCR, polymerase chain reaction
- Ct, cycle threshold
- ESA, epithelial cell specific antigen
Over the past three decades, Western populations have witnessed a 10% year-on-year increase in the incidence of oesophageal adenocarcinoma while the incidence of oesophageal squamous carcinoma remains unchanged.1 Adenocarcinoma of the lower oesophagus and gastro-oesophageal junction now represents the fastest growing cancer in the western world. 1,2 Most recent cancer registry data from the UK regions confirm the rising incidence, with oesophageal carcinoma occurring in 12–18.7 per 100 000 males,3–5 and with adenocarcinomas now outnumbering squamous cell carcinomas.5 Due to the rapidly increasing incidence, this is likely to be an underestimate of the current prevalence.
Barrett’s oesophagus, a columnar metaplasia of the lower oesophageal epithelium, is the strongest known risk factor for the development of oesophageal adenocarcinoma. It is believed that Barrett’s metaplasia progresses through low to high grade dysplasia before developing into adenocarcinoma.6 Patients with Barrett’s oesophagus have an approximate 0.5–2% annual risk of developing this cancer, which is 30–125 times the risk seen in the general population.7 Barrett’s oesophagus is associated with severe gastro-oesophageal reflux disease (GORD)8 but there is much controversy regarding the exact component of the refluxate that is noxious to the oesophageal epithelium. Reflux of acidic gastric contents has clearly been implicated in Barrett’s metaplasia but there is increasing evidence that reflux of duodenal juices is likely to play an important role in malignant development. In animal models, diversion of duodenal contents into the lower oesophagus leads to potent adenocarcinoma formation,9 and in human studies reflux of duodenal contents is associated with increased severity of oesophageal damage.10,11 Bile acids within this refluxate may be crucial, with deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA), frequent constituents of the refluxate,11 being widely studied in cell culture models.
Progression from Barrett’s metaplasia to adenocarcinoma involves deregulation of key cellular processes, including proliferation, apoptosis, and cellular differentiation.12,13 In general these processes are tightly regulated by many factors, including the proto-oncogene c-myc. Dysregulation of c-myc has been observed in many human cancers,14 including those of the stomach and colon, with a role for c-myc in neoplasia being further supported by an increased incidence of carcinomas in c-myc overexpressing transgenic mice.15 The role of c-myc in tumorigenesis however is complex, with its precise role in cell proliferation, growth arrest, and apoptosis dependent on tissue type and environment.16 The gene encoding c-myc is located in the chromosomal region 8q23–24 and recent studies in Barrett’s adenocarcinoma have identified the chromosomal region 8q as being the most frequently observed region of amplification.17 To date however no comprehensive studies of c-myc protein expression in Barrett’s adenocarcinoma have been reported.
The aims of this study were therefore to characterise expression and localisation of the oncogene c-myc in benign Barrett’s metaplasia and oesophageal adenocarcinoma and to investigate its induction by bile acids present in the duodeno-gastro-oesophageal refluxate.
MATERIALS AND METHODS
Patient tissue
Samples for western blotting were obtained from 20 patients with benign long segment (≥3 cm) Barrett’s metaplasia and 20 patients with oesophageal adenocarcinoma. Matched oesophageal squamous, gastric fundal, and duodenal epithelial biopsies were taken as control tissues from each patient. Tissues from an additional 20 patients with Barrett’s metaplasia and 20 patients with oesophageal adenocarcinoma together with squamous control biopsies were also taken for real time polymerase chain reaction (PCR) estimation of c-myc mRNA levels. Histological confirmation of diagnosis was carried out on adjacent biopsies taken from the same site and at the same investigation. All samples in the Barrett’s metaplasia group were from patients with no macroscopic or histological evidence of dysplasia or adenocarcinoma. For western blotting, biopsies were immediately processed and stored at −20°C. For real time PCR, biopsies were immediately processed for RNA extraction. For immunohistochemistry, sections of normal oesophageal squamous epithelium (n=30), normal duodenal epithelium (n=10), normal gastric fundal mucosa (n=10), benign Barrett’s metaplasia (n=30), Barrett’s metaplasia containing low grade dysplasia (n=10), Barrett’s metaplasia containing high grade dysplasia (n=10), and oesophageal adenocarcinoma (n=30) were selected from a large paraffin tissue archive and histological confirmation of pathology carried out prior to study. Local ethics committee approval was granted for the study.
Cell culture
Cell lines derived from oesophageal squamous carcinoma (OE21),18 oesophageal adenocarcinomas (OE33)18 and TE-7,19 and colorectal carcinoma (SW-480) were maintained in growth media (Dulbecco’s modified Eagle’s medium with 10% calf serum+100 units/ml penicillin and 0.1 mg/ml streptomycin). On reaching 70% confluence, cell lines were stimulated as follows: (1) growth media at pH 4; (2) growth media at pH 4 containing 100 μM DCA; (3) growth media at pH 7 containing 100 μM DCA; (4) growth media at pH 4 containing 100 μM CDCA; and (5) growth media at pH 7 containing 100 μM CDCA. Cells grown in growth media at neutral pH were used as controls for all experiments. Cells were either stimulated continuously over a 24 hour time period or pulsed for one minute and washed with growth media and then incubated in growth media for three hours. All experiments were performed in triplicate. The bile acids DCA and CDCA were chosen as they are common constituents of the refluxate11 and are widely studied in cell culture models. A concentration of 100 μM was used as this has been shown to be a physiological concentration and the minimum concentration of bile acid required to induce a maximal c-myc response in our cell culture model system (results not shown). In addition, one minute was chosen as a time interval of stimulation as this is comparable with the average reflux episode.20
Cell proliferation
Cell proliferation was assessed by use of the CellTiter96 AQueous One Solution Cell Proliferation Assay (Promega Corporation, Madison, Wisconsin, USA), a colorimetric assay system for determining the number of viable cells in proliferation. This was performed according to the manufacturer’s instruction. Briefly, 4×104 cells in 0.5 ml of growth medium were plated into each well of a 24 well plate. Twenty four hours later cells were treated according to a regimen, after which 100 μl of CellTiter96 AQueous One Solution Reagent were added to each well and left to incubate for one hour at 37°C followed by absorbance measurements at 490 nm. Fresh growth medium was included as a negative control. Experiments were performed in triplicate.
Real time PCR
RNA was extracted using Trizol reagent (Gibco, Paisley, UK) according to the manufacturer’s instructions. Freshly prepared RNA (1 μg) was reverse transcribed using a cDNA reverse transcription kit (Promega, according to the manufacturer’s instruction). All reactions were performed on a real time PCR machine (PE7700 ABI Prism) using 18S ribosomal RNA as an internal standard (PE Biosystems, Roche, USA). Each reaction was performed in triplicate and contained 900 nM of c-myc specific 5′ (TCAAGAGGTGCCACGTCTCC) and 3′ (TCTTGGCAGCAGGATAGTCCTT) primers, 1× Mastermix (PE Biosystems), 125 nM c-myc specific probe (5′ FAM-CAGCACAACTACGCAGCGCCTCC-TAMRA 3′), 50 nM 18S 5′ and 3′ primers and 200 nM 18S probe (5′ VIC, 3′-TAMRA labelled), and 0.25 μl of cDNA (equivalent to 12.5 ng of reverse transcribed RNA) in a 25 μl reaction. Reactions without cDNA were included as negative controls. Cycle threshold (Ct) values were obtained graphically for c-myc and the 18S internal standard. Gene expression was normalised to the 18S and represented as ΔCt values. For each sample the mean of the three ΔCt values was calculated. Comparison of gene expression between control and treated samples was derived from subtraction of control ΔCt values from treatment ΔCt values to give a ΔΔCt value, and relative gene expression was calculated as 2−ΔΔCt. Relative gene expression was normalised to 1.0 (100%) of controls. All experiments were performed in triplicate. C-myc levels in biopsies of Barrett’s mucosa or adenocarcinoma samples were compared with matched normal squamous controls normalised to a value of 1.
Western blotting
Endoscopic biopsies and established cell lines following stimulation were processed for western blotting as previously described,21 with monoclonal antibodies against c-myc (Santa Cruz, California, USA; 1:1000) and epithelial cell specific antigen (ESA) (Novocastra Laboratories, UK 1:25), the latter utilised for normalisation of epithelial protein loading. HeLa and Cos cell lysates were used as positive and negative controls, respectively. Positive c-myc immunoreactivity in Barrett’s metaplasia and adenocarcinoma samples (already normalised for ESA) were subject to densitometry using the software NIH Image 1.62.
Immunohistochemistry
Immunohistochemistry was performed by the streptavidin-biotin indirect immunoperoxidase method, as previously described,21 with a monoclonal c-myc antibody (Santa Cruz 1:500). Colonic carcinoma sections with or without primary antibody acted as positive and negative controls, respectively.
Immunofluorescence
Cells were fixed in methanol/acetone, blocked (20% goat serum in 1% bovine serum albumin/phosphate buffered saline), and incubated with a monoclonal c-myc antibody (Santa Cruz 1:50) for one hour prior to labelling with FITC goat antimouse (Jackson Immunoresearch, West Grove, Pennsylvania, USA; 1:500). Cells were washed and incubated in 4′, 6-diamidino-3-phenylindole dihydrochloride hydrate (1:10 000) for one minute prior to visualisation. Omission of primary antibody was employed as a negative control.
Statistics
Fisher’s exact test was used to assess significance in c-myc protein and mRNA expression experiments and Mann-Whitney U tests were used in assessing significance in proliferation experiments between pulse/continual stimulated and unstimulated cells, and analysis of densitometry data of western blots. Two tailed tests were employed using SPSS version 9.0 (SPSS Inc, Chicago, USA) and significance was accepted at p<0.05.
RESULTS
Expression of c-myc in Barrett’s metaplasia and oesophageal adenocarcinoma
Western blotting for c-myc revealed no c-myc protein expression in any of the paired oesophageal squamous, gastric, or duodenal control biopsies examined. C-myc expression was found in 18/20 (90%) adenocarcinomas (p<0.05) and 10/20 (50%) Barrett’s metaplasia cases (p<0.05). Figure 1A is a representative western blot for c-myc protein expression, demonstrating higher c-myc protein expression in oesophageal adenocarcinoma compared with Barrett’s metaplasia. Furthermore, all samples positive for c-myc were subject to densitometry, and the average c-myc expression levels in Barrett’s metaplasia compared with adenocarcinoma are represented in fig 1B.
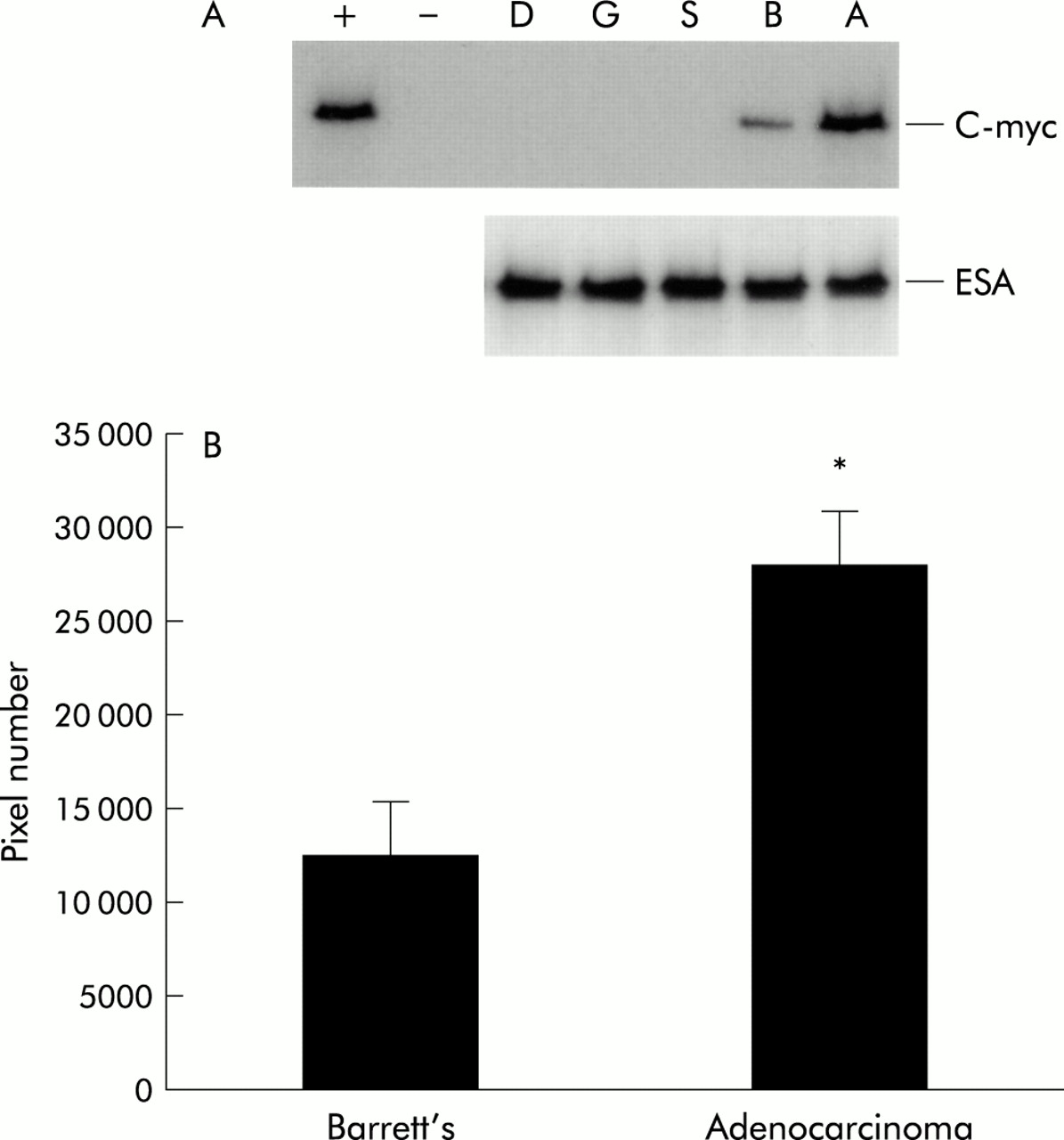
(A) Western blot for c-myc protein demonstrating high c-myc protein expression in oesophageal adenocarcinoma (A) compared with Barrett’s metaplasia (B), with undetectable levels in normal oesophageal squamous (S), gastric (G), or duodenal (D) samples. Equal epithelial protein loading was demonstrated by epithelial cell specific antigen (ESA). HeLa and Cos cell lysates were used as positive (+) and negative (−) controls, respectively. (B) Quantitation of western blots of c-myc by densitometric scanning. All 20 Barrett’s and 20 adenocarcinoma samples were initially normalised for equal epithelial loading with the use of ESA, following which western blots for c-myc were performed. All positive c-myc samples (10/20 in Barrett’s metaplasia group and 18/20 in the adenocarcinoma group) were subsequently subject to densitometry and data are presented as mean (± 2 SEM) pixel number. *Significant difference (p<0.05) between the adenocarcinoma and Barrett’s metaplasia group.
By real time PCR we observed upregulation of c-myc mRNA in 33% of Barrett’s metaplasia cases (p<0.05) and in 84% of oesophageal adenocarcinoma cases (p<0.05) compared with paired control tissue. In those cases that showed upregulation of c-myc, the median fold increase was 2.37 (range 1.29–80) in Barrett’s group and 2.44 (range 1.24–82) in the adenocarcinoma group.
Immunohistochemical analysis of c-myc revealed no immunoreactivity in gastric mucosa (fig 2F) or in duodenal mucosa (results not shown), with very low expression of nuclear c-myc only in the basal layer of normal squamous epithelium (fig 2A). Nuclear c-myc expression was observed in all cases of Barrett’s metaplasia examined, more intense than in normal squamous epithelium (fig 2B). Cytoplasmic immunoreactivity was not seen in any case. In low and high grade dysplasia however, c-myc was not only expressed in the nucleus but there was evidence of diffuse cytoplasmic staining (fig 2C). This cytoplasmic immunoreactivity was further accentuated in all cases of adenocarcinoma (fig 2D, 2E).
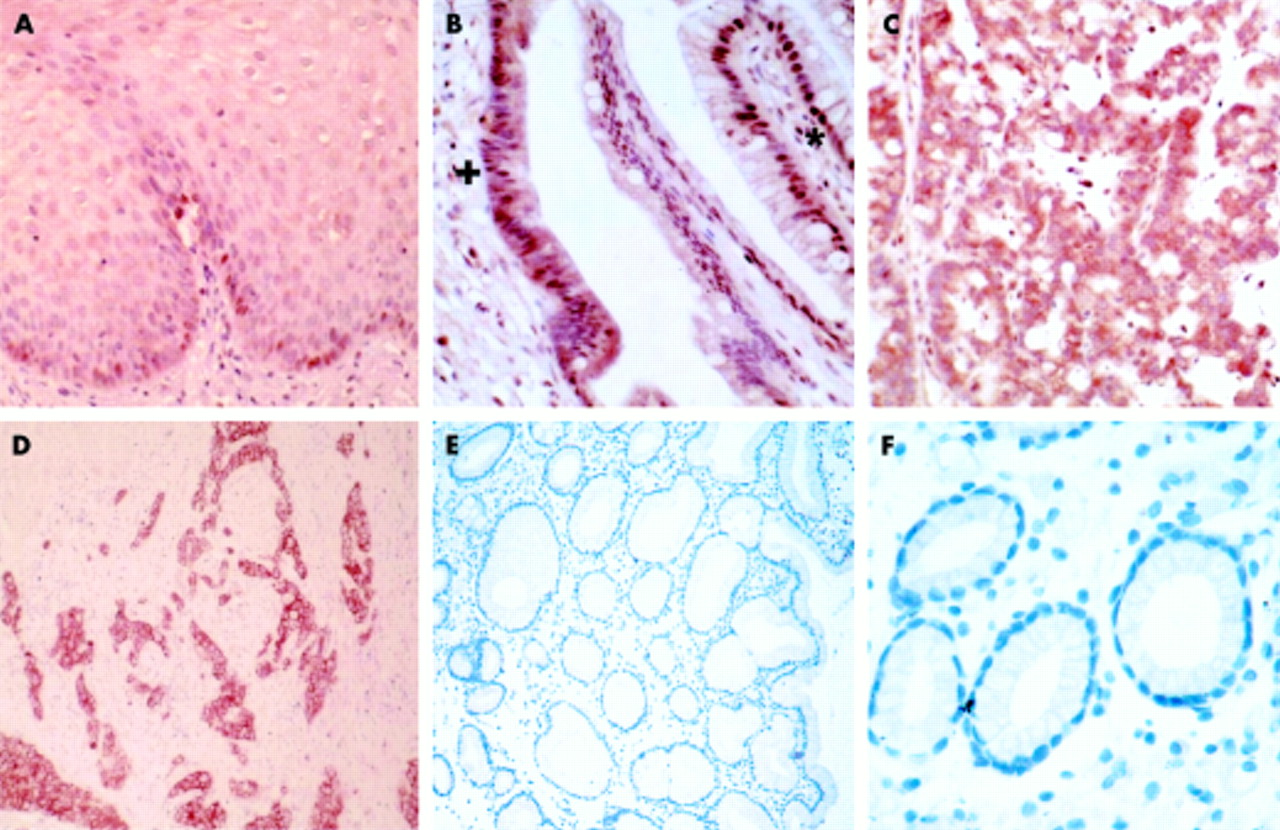
Immunolocalisation of c-myc in the malignant progression of Barrett’s oesophagus (×40 objective), demonstrating localised nuclear staining in the basal layer of the squamous epithelium (A) and in benign metaplasia (B) (denoted by *), becoming cytoplasmic in dysplasia (B) (denoted by +). (C) High grade dysplasia exhibiting both nuclear and cytoplasmic c-myc immunoreactivity, which was further accentuated in adenocarcinoma (D). Adenocarcinoma at lower magnification (E) (×25 objective). No c-myc immunoreactivity was observed in gastric mucosa (F).
Induction of c-myc expression by continuous stimulation with bile acids
To investigate induction of c-myc in the clinical setting of GORD, we investigated the effects of acid and bile acids (at a physiological concentration11) on established cell cultures using the regimens described in materials and methods. The TE-7 cell line derived from a human oesophageal adenocarcinoma was utilised for initial studies. Induction of c-myc was observed in cells stimulated continuously with either DCA at pH 4 or CDCA at pH 4 only. DCA or CDCA at pH 7, or acidified media alone, failed to induce a significant elevation in c-myc over the 24 hour time course examined (fig 3). Increased transcription of c-myc was first detected at 10 hours post-stimulation, with maximal levels observed at 24 hours. C-myc localisation in cells treated with the different regimens was examined by immunofluorescence (fig 3). Only cells treated with DCA or CDCA at pH 4 showed any immunoreactivity, with c-myc most pronounced in and around the nucleus (fig 3B). To extend these observations, the experimental regimens were repeated in the cell lines OE21, OE33, and SW-480. Results of real time PCR analysis and immunofluorescence were virtually identical to those seen in the TE-7 cell line (fig 4). As observed in the TE-7 cell line, c-myc was localised predominately in the nucleus with some perinuclear immunoreactivity. As c-myc has roles in regulating proliferation, we assessed the levels of proliferation in these 24 hour stimulated cells (fig 5). Cells in continuous culture in the presence of both bile and acid caused a dramatic reduction in proliferation compared with control unstimulated cells (p<0.05). However, this suppression in proliferation was also observed when cells were challenged with acid alone (p<0.05), while bile acids alone (pH 7) had no effect on the rate of proliferation compared with cells in growth media alone.
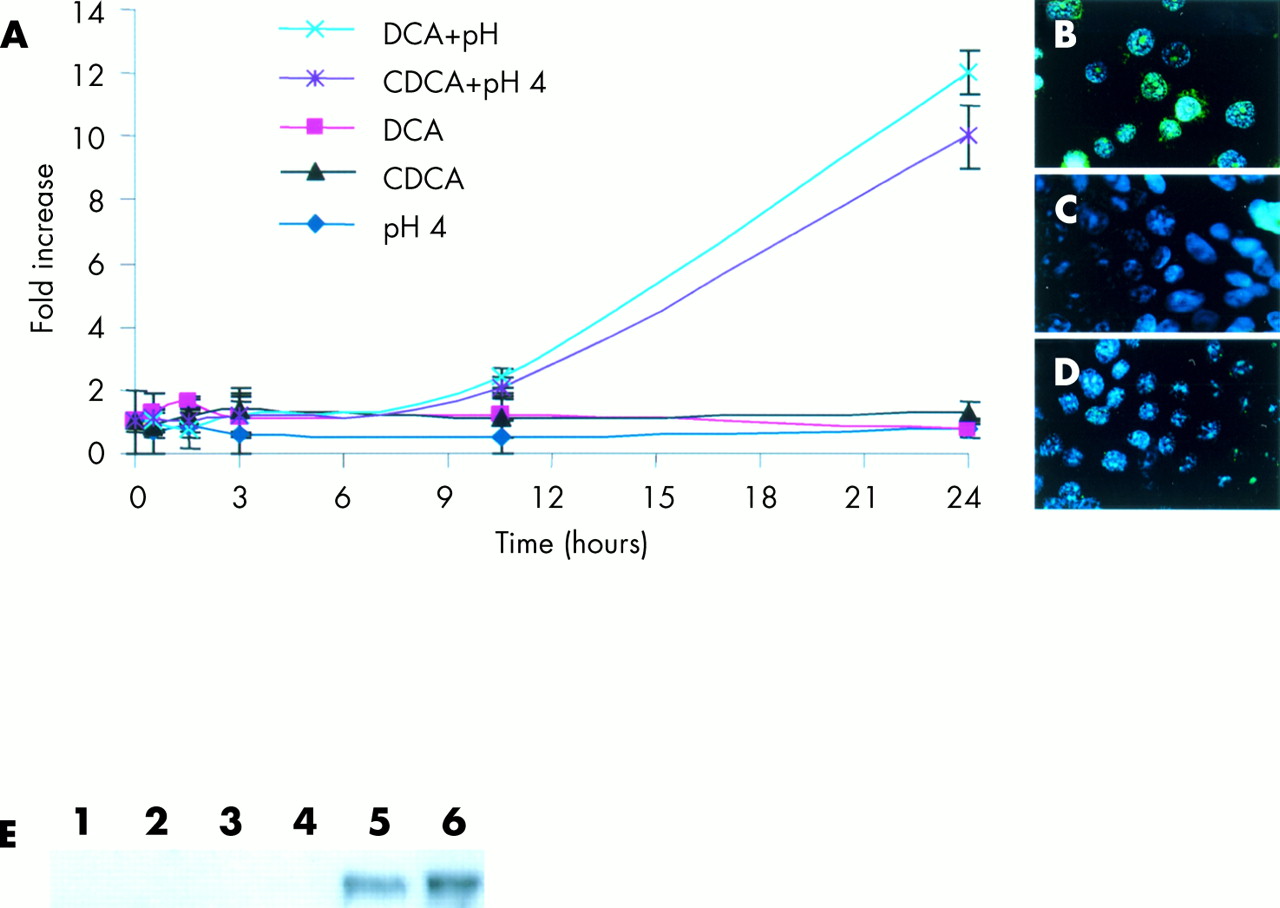
C-myc mRNA levels in TE-7 cells over a 24 hour stimulation period (A). Induction of c-myc in cells stimulated with deoxycholic acid (DCA) at pH 4 or chenodeoxycholic acid (CDCA) at pH 4, but not in cells stimulated by DCA or CDCA at pH 7 or acidified (pH 4) growth media alone. Data are the mean (±2 SEM) of three experiments. Immunofluorescence of c-myc (FITC) in TE-7 cells stimulated by DCA at pH 4 or CDCA at pH 4 (B), DCA or CDCA at pH 7, or acidified growth media (pH 4) alone (C) and unstimulated cells (D) (×60 objective). (E) C-myc protein expression by western blotting. Lane 1, TE-7 cells cultured in growth media alone; lane 2, pH 4; lane 3, DCA (pH 7); lane 4, CDCA (pH 7); lane 5, DCA (pH 4); and lane 6, CDCA (pH 4). HeLa and Cos cell lysates were used as positive (+) and negative (−) controls, respectively. Cell lysates were normalised for equal protein loading by Coomassie blue staining.

Stimulation of epithelial cell lines SW-480, OE33, and OE21 with deoxycholic acid (DCA) (pH 4) or chenodeoxycholic acid (CDCA) (pH 4) resulted in a marked induction in c-myc mRNA over a 24 hour time period in all cell lines. Data are the mean (±2 SEM) of three experiments. Induction of c -myc protein at 24 hours was confirmed by immunofluorescence (FITC) in DCA (pH 4) or CDCA (pH 4) stimulated (B) compared with unstimulated (A) cells (×60 objective).
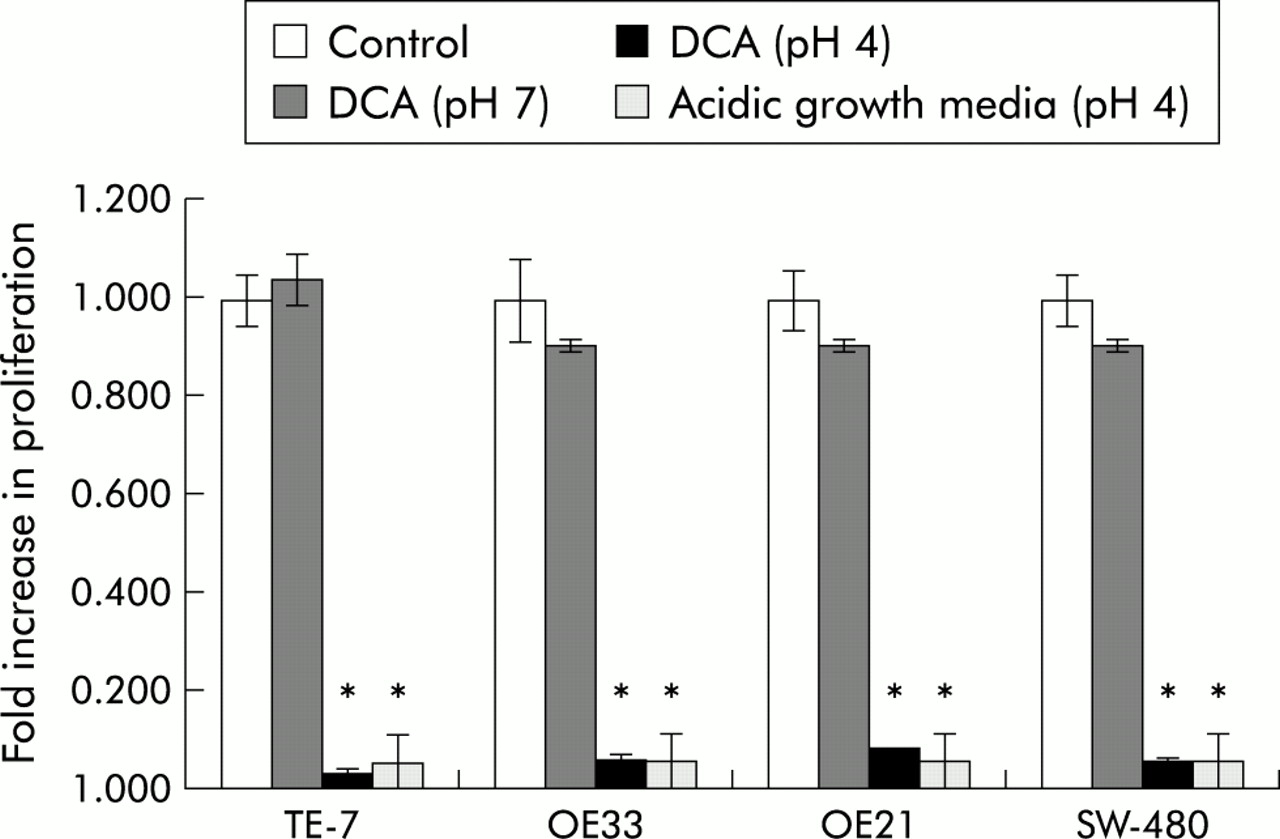
Proliferation levels assessed in TE-7, SW-480, OE33, and OE21 cell lines following 24 hour exposure to deoxycholic acid (DCA) (pH 7), DCA (pH 4), acidic growth media (pH 4) compared with cells cultured in growth media alone (control). Data are the mean (±2 SEM) of three experiments. *Significant difference (p<0.05) between stimulated and unstimulated cells.
Effect of pulsed DCA and CDCA (pH 4) on induction of c-myc
As GORD is most commonly an intermittent event, characterised by repeated short exposures to refluxed juices, we attempted to mimic this by stimulating the four cell lines (OE21, OE33, TE-7, and SW-480) according to the previously described regimens for one minute only, followed by replacement with growth media for three hours. One minute exposure of bile acid followed by a three hour incubation in growth media was sufficient to induce c-myc mRNA by 1.7–3.4 fold in the four cell lines tested (p<0.05) (fig 6). Acid alone or bile acids at pH 7 once again failed to induce c-myc (results not shown). Induction of c-myc was associated with a 15–22% (p<0.05) increase in proliferation in the four cell lines compared with unstimulated cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Stimulation of epithelial cell lines SW-480, TE-7, OE33, and OE21 with a one minute pulse of deoxycholic acid (DCA) (pH 4) followed by a three hour incubation in growth media induced a marked induction in c-myc mRNA compared with unstimulated cells. (B) Proliferation levels in all four cell lines following a one minute pulse of DCA (pH 4) followed by a three hour incubation in growth media compared with unstimulated cells. Data are the mean (±2 SEM) of three experiments. *Significant difference (p<0.05) between stimulated and unstimulated cells.
DISCUSSION
The proto oncogene c-myc has been implicated in many human cancers,14 including those of the stomach and colon. To our knowledge we are the first to identify upregulation of c-myc protein and mRNA in Barrett’s metaplasia and oesophageal adenocarcinoma. Western blotting demonstrated upregulation of c-myc in 50% of benign Barrett’s metaplasia and 90% of oesophageal adenocarcinoma samples studied. In addition, densitometry of western blots revealed significantly more c-myc in adenocarcinoma than in Barrett’s metaplasia samples. At the RNA level it was apparent that 33% of Barrett’s showed upregulation of c-myc compared with 84% in adenocarcinoma samples, comparable with what is observed at the protein level. Interestingly, the median increase in c-myc mRNA between the two groups was identical. This would suggest that this further increase in c-myc protein expression observed in adenocarcinoma compared with Barrett’s metaplasia is likely to be as a consequence of post translational mechanisms which may increase stability or decrease turnover of c-myc.
We have also shown aberrant c-myc localisation at progressive stages in the metaplasia-adenocarcinoma sequence. Discrete nuclear staining for c-myc was observed in benign Barrett’s columnar metaplasia. As the epithelium became dysplastic, expression of c-myc within the cytoplasm in addition to nuclear staining was apparent; this aberrant localisation was further accentuated in adenocarcinoma. In other malignancies it has been suggested that this aberrant localisation may be due to alterations in the C terminus of the protein, reducing the efficiency of nuclear targeting.22 Moreover, if this additional cytoplasmic pool of c-myc has a dissimilar turnover/stability to the nuclear pool, this could explain the discrepancy between the median fold mRNA increases and protein levels.
These observations of elevated c-myc are supported by a recent study examining chromosomal imbalances in Barrett’s adenocarcinoma where it was reported that the minimal common chromosomal region 8q23–24, which contains the c-myc sequence, was the most common region amplified, with amplifications occurring in 80% of cases examined.17
As c-myc over expression was very common in samples of adenocarcinoma (90%), this would suggest that it is likely to be one of the many events in the development of adenocarcinoma. However, because a high proportion of patients with benign Barrett’s metaplasia have dysregulated c-myc (50%) it is unlikely that c-myc is sufficient alone to cause progression to adenocarcinoma as patients with Barrett’s metaplasia have only a 0.5–2% annual risk of developing this cancer.7
As well as c-myc being important in cellular proliferation, it has a role in sensitising cells to apoptotic triggers. Such a role however may be abrogated by aberrations in downstream apoptotic control mechanisms such as p53 mutations, frequently observed in oesophageal adenocarcinomas.23 Loss of apoptosis could then leave the increased proliferation unchecked and contribute to malignant progression, a potential mechanism whereby dysregulated c-myc in Barrett’s metaplasia might have an increased risk of malignant progression.
Reflux of juices from the stomach and duodenum has been strongly associated with adenocarcinoma of the oesophagus, and we therefore investigated components of this refluxate as potential c-myc inducers. The unconjugated bile acids DCA and CDCA have been shown to be frequent components in duodenal gastro-oesophageal reflux,11 and have been widely studied in cell culture models. Although bile acids are normally conjugated prior to excretion by the liver, deconjugation frequently occurs in the gastrointestinal tract under the action of certain intestinal bacteria.24
Our studies showed potent induction of c-myc by both DCA and CDCA but only in acidic conditions, and that bile acids at neutral pH were unable to induce c-myc in a 24 hour continual culture system. This could be explained by the predominance of the ionised forms of the bile acids at pH 7, which cannot penetrate the phospholipid bilayer. At pH 4 bile acids are non-ionised and can penetrate the cell membrane. Once inside the cell the bile acids become reionised and are unable to exit the cell, leading to a gradual intracellular accumulation of ionised bile acids. This intracellular accumulation of bile acids at low pH has been supported by other studies in the stomach and oesophagus.25 Why the magnitude of c-myc mRNA induction was variable between the four cell lines is not known although it is possibly due to differences in c-myc regulatory proteins. Furthermore, cells challenged with acidic growth media alone were unable to induce c-myc. However, it was of interest that 24 hour continuous culture with bile acids at pH 4 caused complete suppression of proliferation. This is likely to be due to the acidic milieu as acid alone on these cells caused a dramatic loss of proliferation, consistent with previous reports.26 This suggests that in a continual culture system, bile acid can induce c-myc, but any increased proliferation is likely to be overridden by the acid mediated suppression of proliferation.
In patients with GORD, reflux is inherently an intermittent event,20,27 and we attempted to mimic this in vitro. In all four cell lines tested, a single one minute pulse of acidified DCA or CDCA followed by three hours in growth media led to a marked induction in c-myc mRNA. More importantly, unlike continuous bile acid exposure, pulsing in this fashion caused a modest induction of proliferation. Hence it appears that acidified bile can induce c-myc with a resulting increase in cellular proliferation. Our data are in contrast with Kaur et al who have demonstrated that a one hour pulse of a bile acid mixture caused suppression of proliferation.28 However, caution must be exercised in comparing data from these two studies as there were a number of experimental differences, including bile acids used, concentrations, and model systems, with probably the most important difference being duration of stimulation. What can be inferred from both of these studies is that duration and pattern of bile acid exposure may be critical in terms of biological response.
The precise mechanism of bile acid mediated proliferation observed in this study is likely to be a highly complex event involving a cascade of signalling proteins and pathways. Several elegant studies have shown that acid mediated proliferation involves factors such as activation of the Na+/H+ exchanger,29 protein kinase C,29 and the mitogen activated protein kinase pathways30 while bile salt induced proliferation is mediated by protein kinase C.28 Further work is required to delineate whether these pathways are instrumental in regulating bile acid mediated proliferation observed in this study.
If our in vitro findings were representative of an in vivo setting such as Barrett’s metaplasia then it would appear that control of bile reflux into the oesophagus could potentially reduce expression of c-myc. While it might be argued that antireflux surgery would be the best way of achieving this aim, we have shown that bile acids upregulate c-myc only in an acidic environment and hence acid suppression by proton pump inhibition or other means could equally prevent induction. Clearly, this would have major implications for chemopreventative strategies for Barrett’s adenocarcinoma.
While induction of c-myc by environmental cues such as refluxate is likely to be important, it is certainly not the only means of induction of this oncogene, and there are likely to be many other mechanisms of c-myc induction such as by gene amplification.
In summary, we have shown upregulation of c-myc protein in 50% of Barrett’s metaplasia cases and 90% of oesophageal adenocarcinomas, with upregulation also being confirmed at the mRNA level. We have also identified a role for bile acids in an acid environment as potent inducers of this oncogene. The resulting effects of c-myc are likely to be complex but it is clear that a series of factors, including duration and pattern of reflux, genetic predisposition, and environmental influencers are likely to be critical in determining not only these effects but also expression of c-myc itself. The in vitro studies were carried out in cell lines derived from malignant tumours and it remains to be proved whether these mechanisms are equally as important in non-malignant tissues. However, the finding that c-myc expression is elevated in most oesophageal adenocarcinomas and in 50% of Barrett’s metaplasias, and that it can be induced by bile acids in acidic conditions, could help to explain the association between reflux of duodenal juices and oesophageal adenocarcinoma.
Acknowledgments
This study was funded by the Cancer Research Campaign, UK, and the Royal College of Surgeons of England, UK.