Article Text

Abstract
Background and aims: Large numbers of plasma cells (PC) localise in the intestinal lamina propria (LP) where they play a critical role in the defence against pathogens. This study analyses the level of maturation reached by normal human colon LPPC in comparison with that of bone marrow (BM) PC.
Methods: A technique was designed to purify LPPC by combining collagenase digestion of the mucosal layer and immunomagnetic selection of CD54+ LP cells. It provided highly purified PC, as demonstrated by morphology, CD38h phenotype, and cytoplasmic IgA staining criteria. This procedure allowed comparison of in vitro functional capacities and a broad phenotypic analysis of BMPC and LPPC.
Results: LPPC and BMPC exhibited identical expression of differentiation markers (CD19−/+, CD20−, HLA-DRlow/−, VS38chigh), survival molecules (CD95 low/−, Bcl-2+), and B cell transcription factor profile, as well as similar in vitro Ig secreting kinetics (14 days) and lack of susceptibility to apoptosis by CD95 ligation. In contrast, they markedly differed in adhesion molecule expression, as LPPC showed higher levels of CD44 and CD21 and were α4β7+ whereas BMPC lacked this integrin and expressed higher levels of CD49d and CD31.
Conclusion: These data indicate that PC at effector sites of the humoral response (BM and LP) show similar high differentiation, survival, and functional features but display a distinctive pattern of adhesion molecules, probably related to their respective homing locations.
- colon
- lamina propria plasma cells
- phenotype
- transcription factor expression
- adhesion molecule profile
- Ab, antibody
- Blimp-1, B lymphocyte induced maturation protein 1
- BM, bone marrow
- BSAP, B cell specific activation protein
- CyC, Cy-Chrome
- FITC, fluorescein isothiocyanate
- LP, lamina propria
- mAb, monoclonal antibody
- MFI, mean fluorescence intensity
- PBS, phosphate buffered saline
- PC, plasma cells
- PE, phycoerythrin
- RT-PCR, reverse transcriptase-polymerase chain reaction
Statistics from Altmetric.com
- Ab, antibody
- Blimp-1, B lymphocyte induced maturation protein 1
- BM, bone marrow
- BSAP, B cell specific activation protein
- CyC, Cy-Chrome
- FITC, fluorescein isothiocyanate
- LP, lamina propria
- mAb, monoclonal antibody
- MFI, mean fluorescence intensity
- PBS, phosphate buffered saline
- PC, plasma cells
- PE, phycoerythrin
- RT-PCR, reverse transcriptase-polymerase chain reaction
Plasma cells (PC) are considered the final stage of the B lymphocyte differentiation process and as such show features that reveal their full specialisation in the synthesis and secretion of antibodies (Ab). Therefore, PC are ultimately responsible for the formation of Ab in response to antigens and for adequate maintenance of the Ab level of the organism. In animal models of systemic humoral immune responses, specific PC initially appear in antigen activated foci and germinal centres of inductive lymphoid organs (lymph nodes, spleen).1–3 Subsequently, some of these cells are thought to migrate into the bone marrow (BM), the final deposit and effector organ for PC elicited in systemic responses.4,5 Human BM also becomes the final reservoir of PC formed in distant inductive territories following systemic antigen stimulation and, accordingly, human BMPC generate the majority of serum Ig formation.6 Although PC present in different organs show a similar morphology and Ig secreting role, evidence increasingly demonstrates the existence of relevant differences within the PC compartment. In rodent models, BMPC show a prolonged lifespan (from several weeks to several months) while the majority of PC present in inductive organs die in 2–3 days,7–9 apparently by apoptosis.10 In addition, PC exhibiting IgVH gene somatic mutations that confer higher affinity for the antigen are progressively enriched in the BMPC pool during the course of the response.11,12 In humans, PC occurring in peripheral lymphoid organs such as lymph nodes, blood, and tonsils show short term kinetics of Ig secretion in vitro13–16 and exhibit a propensity to undergo apoptosis either spontaneously or induced by CD95 cross linking17–19; in contrast, human BMPC produce Ig in vitro for prolonged periods of time (at least two weeks), and are not susceptible to apoptosis through ligation of the death receptor CD95.18,20,21 High expression of the CD38 molecule (CD38h phenotype) by all human PC22 has provided a tool for comparative phenotypic analysis of PC obtained from different territories. It reveals that BMPC exhibit features indicating a level of maturation higher than that shown by PC from inductive areas (tonsils, blood).23 Taken together, these findings support the view that PC present in the BM, the effector site for systemic humoral responses, have reached the highest survival state and functional capacities. The mechanisms that underlie migratory and maturative events giving rise to BMPC remain to be elucidated.
Less attention has generally been paid to PC responsible for the mucosal humoral immune response. Nevertheless, the number of PC generated by mucosal antigen stimulation is larger and they produce more Ig than PC induced in systemic responses.24,25 Thus following the arrival of antigens at a variety of mucosal associated lymphoid organs, antigen induced foci and activated germinal centres are rapidly developed.26 Examples of such organs are intestinal Peyer’s patches and, in part, the tonsils, which collectively are considered inductive areas for mucosal humoral immune responses. It is thought that from these inductive areas antigen primed B cells migrate through the circulation into effector sites for mucosal humoral responses, the most prominent of which is the intestinal lamina propria (LP), where large numbers of PC committed to mucosal antigen responses are commonly found.27 These PC predominantly produce Ab of the IgA isotype which are mainly secreted across mucous membranes and whose function is essential for defence against the arrival of potentially invasive micro-organism at the mucosal surfaces.28 Recent experiments in mice show that maturation of certain intestinal IgA secreting PC, mostly committed to commensal bacteria, can occur from B1 lymphocytes, and these PC follow a T cell and lymphoid follicle independent pathway, apparently guided by interaction with LP stromal cells.29,30 Evidence supporting this pathway in humans has not as yet been documented. In fact, the majority of human LPPC harbour heavily mutated IgV genes,31 indicating that they have undergone strong antigen selection, a feature apparently restricted to the germinal centre pathway and characteristic of highly differentiated PC.11,12 Despite their importance, a wide phenotypic analysis of LPPC has not as yet been performed. In particular, it is not known whether LPPC share similarities with PC from the BM, the other well established PC effector site. Such a study has been hampered by the lack of a suitable method for LPPC isolation.
The present work reports a fast and effective method for purifying PC from human colon LP. This technique allowed us to confirm the correspondence between LP CD38h cells and PC and to explore “ex vivo” functional capabilities of isolated LPPC, such as the kinetics of Ig secretion and CD95 ligation effect. In order to gain a deeper insight into the maturational stage of LPPC, their expression of several molecules was explored using labelled mouse monoclonal antibodies and flow cytometry analysis. These molecules include differentiation markers, and survival related and adhesion molecules. In addition, the presence of mRNA for the two relevant B cell transcription factors B lymphocyte induced maturation protein 1 (Blimp-1) and B cell specific activating protein (BSAP) was investigated. The results were compared with those of other PC populations, particularly those from BM.
MATERIALS AND METHODS
Materials
Cycloheximide, purified mouse IgG, and collagenase V were purchased from Sigma (St Louis, Missouri, USA). EDTA was provided by Pharmacia Biotech (Uppsala, Sweden). Purified mouse monoclonal antibody (mAb) against CD54, fluorescein isothiocyanate (FITC) labelled mAb against CD19 and CD44, phycoerythrin (PE) labelled mAb against CD19, CD20, CD31, CD49d, CD54, CD95, HLA-DR, and mouse IgG, Cy-Chrome (CyC) labelled mAb against CD38, the corresponding isotypic negative controls, and PE labelled rat antimouse IgG mAb were purchased from Becton Dickinson (San Jose, California, USA). Apoptosis inducing mAb against CD95 (clone CH11) and PE labelled mAb against CD21 were provided by Coulter (Miami, Florida, USA). FITC labelled mAb against VS38c and Bcl-2, FITC conjugated rabbit antihuman IgA, and the intrastain fixation permeabilisation kit were from Dako (Glostrup, Denmark). Unconjugated and peroxidase conjugated goat F(ab)’2 antihuman IgA and anti-human IgG used in ELISA were purchased from Biosource (Camarillo, California, USA). ACT-1 mAb against the adhesion molecule α4β7 was kindly provided by Dr Ortíz de Landázuri (Hospital de la Princesa, Madrid, Spain). Goat antimouse IgG bound to magnetic microbeads, selection columns of LS+ type, and midiMACS magnet were obtained from Miltenyi Biotec (Auburn, California, USA). Ninety six well flat bottomed culture plates and U96 Maxisorp plates used for ELISA were provided by Nunc (Roskilde, Denmark)
Isolation of colon lamina propria (LP), tonsil, and bone marrow (BM) cells
Human colonic tissue (fig 1A, B) was obtained from normal areas of surgically resected specimens. The mucosal layer was mechanically dissected out (fig 1C) from the colon wall sample using two scalpels. This process released a fine tissular sheet including the epithelium and LP (fig 1D). After washing in phosphate buffered saline (PBS), the layer was minced in a Petri dish and treated with collagenase V (1 mg/ml in RPMI-1640) for 15 minutes at 37° C in a shaking bath. The reaction was stopped by adding culture medium (see below), and the resulting cell suspension was centrifuged at low speed (20 g, three minutes) to separate undigested tissue pieces. The supernatant containing released cells was collected, centrifuged at 400 g for seven minutes, and washed twice in culture medium. The resulting cell fraction will be referred as to LP cells. BM and tonsil PC were purified as previously reported.23 Approval was obtained from the Institutional Review Board (Comité Etico, Hospital Universitario Puerta del Mar) for these studies.

Purification of plasma cells (PC) from human colon lamina propria (LP). The figure shows a representative example of the consecutive steps followed in the protocol used for the purification of LPPC. (A) Haematoxylin-eosin staining of a colon wall section showing the different layers. (B) Giemsa staining of the LP area showing abundant PC. (C, D) Haematoxylin-eosin staining of the dissected mucosal layer (including epithelium and LP areas) and the remaining tissue, respectively. (E) LP cells were obtained by collagenase digestion of the mucosal layer (C) and studied using labelled monoclonal antibodies (mAb) and flow cytometry analysis. A dot plot analysis of the CD19/CD38 cell expression is shown: a cell subset of CD38h CD19+/− can be observed. (F) Dot plot analysis of LP cells labelled for CD38 and CD54 showing the distinctive expression of CD54 by the LP CD38h cells. (G) Dot plot analysis of CD38 and CD19 expression of LP CD54 selected cells showing the high degree of enrichment in CD38h cells obtained by this procedure. (H, I) CD38h cells purified by CD54 immunomagnetic selection were identified as PC by Giemsa staining as well as by intracytoplasmic IgA staining of cytospin preparations, respectively.
Isolation of CD54+ cells from LP cell preparations
CD54+ cells were purified from the LP cell fraction by an immunomagnetic technique. Briefly, cells (up to 100×106 cells/ml) were incubated with anti-CD54 mAb (10 μg/ml) in 2 mM EDTA 0.5% bovine serum albumin in PBS for 10 minutes in the dark at 4° C. After two washes in the same buffer, cells (up to 100×106 cells/ml) were incubated with goat antimouse magnetic micro beads, according to the manufacturer’s instructions. After two washes, cells were resuspended (up to 50×106 cell/ml) in the buffer. A separation column was placed in the magnet and 500 μl of buffer were applied at the top of the column and allowed to run through, the effluent being discarded. The cell suspension was pipetted onto the column and allowed to run through. The effluent was collected as negative cell fraction. The column was washed three times with 3 ml of buffer and the effluents included in the negative cell fraction. At this point, the column was removed from the magnet, 5 ml of buffer were applied at its top, and cells were firmly flushed out, using the supplied plunger. The effluent was collected as the positive cell fraction (LP CD54+ cells).
Cell culture and IgA ELISA
LP CD54+ cells were adjusted to 0.5×106 cells/ml in a culture medium consisting of RPMI 1640 supplemented with 10% fetal calf serum, l-glutamine (10 mM), and gentamycin (0.05 mg/ml), and were cultured in 96 well plates in a final volume of 250 μl/well at 37° C with 5% CO2. In some experiments cells were cultured in the presence of the apoptosis inducing anti-CD95 mAb CH11 at 400 ng/ml. After four, seven, and 14 days, cell free supernatants were collected, and IgA secretion was tested by enzyme linked immunoabsorbent assay in microtitre plates, as previously reported.16
Cell staining and flow cytometry
For three colour labelling experiments, 200 μl of LP and BM cells (at 5×106 cell/ml) were incubated with optimal concentrations of a variety of labelled mAb (see below) for 20 minutes in the dark at 4° C. After two washes, cell phenotype was analysed by flow cytometry. To detect Bcl-2 and VS38c (both intracellular molecules), a fixation/permeabilisation kit (Dako) was used after surface staining, following the manufacturer’s instructions. α4β7 expression was detected by a direct/indirect staining technique. Cells were incubated with mouse ascites containing ACT-1 mAb (1/100 dilution), washed, and stained with a PE conjugated rat antimouse mAb. After washing, cells were incubated with purified mouse IgG (1 μg/ml) for 30 minutes to block any free binding site of the secondary mAb. After two washes in PBS, cells were stained with FITC labelled mAb against CD19 and CyC labelled mAb against CD38. All incubation and washing steps were performed under the conditions indicated above. FACS analysis was performed on a FACScalibur cytometer (Becton Dickinson) equipped with an air cooled argon ion laser emitting 15 mW at 488 nm. The instrument was equipped with three fluorescence detector photomultiplier tubes, with green fluorescence (FITC) being collected through a 530/30 nm bandpass, red/orange (PE) through a 585/42 nm bandpass, and red (CyC) through a 650 nm longpass filter. Cell analysis was performed with Cellquest software (Becton Dickinson). Light scatter signals were recorded in linear mode and fluorescence signals in logarithmic mode. PC present in the LP and BM mononuclear cell fractions were identified by their CD38h expression profile in a CD19/CD38 dot plot. The third fluorescence was used to explore PC expression of differentiation markers (CD19, CD20, VS38c, and HLA-DR), survival factors (CD95 and Bcl-2), and adhesion molecules (CD21, CD31, CD44, CD49d, CD54, and α4β7). Data from 2000–5000 CD38h cells/sample were collected, and the percentage as well as mean fluorescence intensity (MFI) of CD38h cells positive for each analysed molecule were monitored. Results are expressed as mean (SEM) of a variety of experiments. Statistical analysis was carried out by using the Student’s t test. Differences were considered significant when p<0.05.
Detection of the B cell transcription factors Blimp-1 and BSAP
The presence of transcripts of Blimp-1 and BSAP was investigated in human highly purified PC from tonsils (T-PC), LP (LPPC), and BM (BMPC) by reverse transcriptase-polymerase chain reaction (RT-PCR). To this end, total RNA from each cellular fraction was purified using the acid-guanidine-thiocyanate-phenol-chloroform method.
After a DNAse I treatment, first strand cDNA copies were synthesised using avian myeloblastosis virus reverse transcriptase (Promega, Barcelona, Spain) with random hexamers (pd(N6); Amersham Pharmacia Biotech, Barcelona, Spain) for priming. Then, PCR was performed with the following oligonucleotide primers: for Blimp-1 sense primer 5′-ATGCGGATATGAC TCTGTGGA-3′ and antisense primer 5′-CTCGGTTGCTTTAGACTGCTC-3′, for BSAP (sense) 5′-CAGCATAGTGTCCACTGGCT-3′ and (antisense) 5′-CCTGTCAGCGTCGGTGCTGA-3′. cDNA (3 μl) was amplified in a PTC-100 MJ Thermocycler (MJ-Research Inc., Waltham, Massachusetts, USA) using each primer and Taq DNA polymerase (Biotaq, Bioline Ltd, UK). The cycler conditions for Blimp-1 and BSAP consisted of a denaturing step at 94°C for one minute, an annealing step at 65°C for one minute, and an amplification step at 72°C for one minute, for 35 cycles, and a final additional amplification step at 72°C for seven minutes. The amplified products were analysed on a 1.2% agarose gel containing ethidium bromide and visualised by UV light illumination. The quantity of β-actin cDNA was evaluated using a sense primer 5′-TACCACTGGCAT CGTGATGGACT-3′ and antisense primer 5′-CGTCACACTTCATGATGGAG-3′ as cDNA internal controls. The cycler conditions for β-actin were as above, except that the annealing temperature was 62°C and only 30 cycles were performed.
Histology and plasma cell identification
Whole colon tissue and fragments resulting from dissecting out the mucosal layer were processed for histological analysis. In brief, samples were formalin fixed, paraffin embedded, and stained with haematoxylin and eosin. Whole colon tissue slides were additionally stained by a Giemsa technique to identify PC “in situ”. LP CD54 cells (0.1×106 cells in 100 μl of PBS) were cytocentrifuged on slides and also stained (Giemsa), and cells with PC morphology were determined. Cells containing intracytoplasmic IgA were detected on similar cytospin cell preparations by a direct immunofluorescence technique. Positive cells were identified by fluorescence microscopy.
RESULTS
Purification of human colon lamina propria plasma cells (LPPC)
The first aim of this study was to design a protocol to purify human LPPC in a period of time that preserved PC and allowed their functional assessment. A colonic mucosal layer that included epithelium and LP (fig 1C) was obtained and a cell suspension was released after a brief collagenase treatment (LP cell fraction). The presence of PC in this cell preparation was investigated by cell staining with labelled mAb, followed by flow cytometry analysis. It is well established that human PC express high levels of CD38 molecules on their surface (CD38h).17–22 As can be seen in fig 1E, a subset of LP cells exhibiting the phenotype CD38h CD19+/− could be clearly detected in the CD38/CD19 dot plot. This cell subset accounted for 21.5 (2)% (mean (SEM); n=16) of the total LP cell fraction which, in addition to this putative PC population, consisted of B and T lymphocytes and epithelial cells (data not shown). Subsequently, LP cells were triple stained (for CD38, CD19, and a third molecule) in search of a marker present on CD38h cells that was suitable for use in an immunomagnetic purification strategy. Among all the molecules tested, CD54 was the co-expressed marker that permitted the clearest distinction of CD38h cells from the rest of the LP cells (fig 1F). Accordingly, immunomagnetic selection by the presence of CD54 was carried out in LP cell populations. Selected LP CD54+ cells were all positive for expression of the CD54 molecule, as expected (data not shown). Subsequently, the LP CD54+ cell fraction was stained for CD38 and CD19 showing a highly purified CD38h population (fig 1G). CD54+ selected cells consisted almost entirely of CD38h cells (94.4 (1)%; mean (SEM); n=4). In addition, LP CD54+ selected cells contained intracytoplasmic IgA (fig 1I) and could be morphologically identified as typical PC (fig 1H). LPPC purification by a direct CD38+ immunomagnetic selection procedure yielded significantly inferior results (data not shown).
“Ex vivo” spontaneous Ig secretion by isolated LPPC: kinetics and anti-CD95 mAb effect
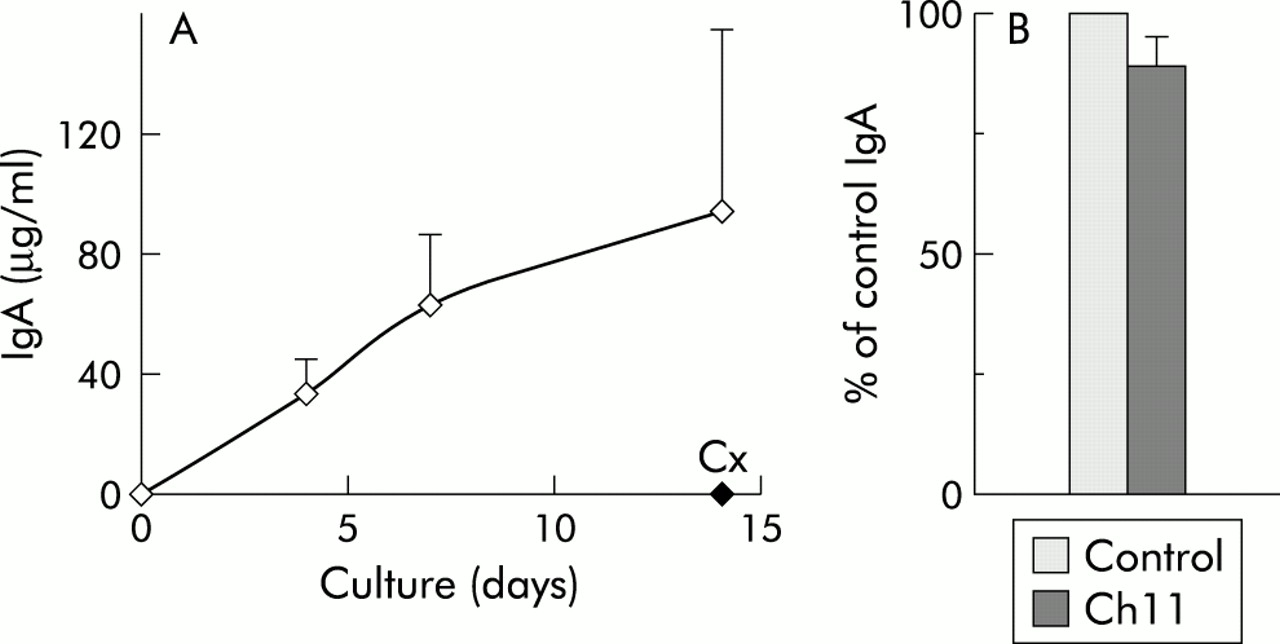
In vitro Ig secreting capacity of LPPC was examined next. Isolated LPPC were cultured for four, seven, and 14 days, culture supernatants were harvested at the end of the culture period, and IgA secretion was quantified by ELISA. As shown in fig 2A, LPPC linearly secreted IgA for 14 days. IgA secretion was actively carried out by the purified LPPC as addition of the protein synthesis inhibitor cycloheximide reduced substantially this capacity. In addition, inclusion of the apoptosis inducing anti-CD95 mAb CH11 into LPPC cultures did not provoke a significant decrease in their spontaneous IgA secretion (fig 2B).

In vitro IgA secretion by lamina propria (LP) CD38h cells. (A) Purified LP CD38h cells were cultured for the indicated periods of time, and the quantity of IgA secreted into the supernatant was assessed by enzyme linked immunoabsorbent assay. Cycloheximide (Cx 10 μg/ml) was added to certain cultures. Results are expressed as μg/ml of IgA, and represent the mean (SEM) of four experiments. (B) LP CD38h cells were cultured in the absence (Control) and presence (Ch11) of the anti-CD95 mAb CH11 (400 ng/ml) for seven days, and IgA secreted into the supernatant was determined. The results are expressed as the percentage of control IgA secretion, and represent the mean (SEM) of six experiments.
Comparative analysis of differentiation markers, death/survival molecules, and B cell transcription factor expression by PC from LP and BM
Expression of several B cell differentiation markers (CD19, CD20, HLA-DR, and VS38c), the death receptor CD95, and the antiapoptotic molecule Bcl-2 was assessed by using labelled mAb and flow cytometry analysis in PC populations from both BM and LP. BM mononuclear cell and LP cell populations were triple stained, and PC were identified as CD38h cells in a CD38/CD19 dot plot. Figure 3 compares expression of these molecules by PC from LP and BM, showing a representative example of the obtained histograms (fig 3A), and a summary of several experiments expressed as percentage of positive PC for each molecule (fig 3B). As can be seen, all differentiation markers were similarly expressed by PC from both LP and BM. Thus CD19 showed a double population (negative/positive), CD20 was completely negative, HLA-DR exhibited a broad spectrum of intensity levels ranging from intermediate to negative expression, and VS38c, an endoplasmic reticulum protein described as a good marker for PC,32 was highly expressed by both PC populations. For expression of the death receptor CD95, there was a mixture of positive and negative PC in both territories. In contrast, BMPC and LPPC were clearly positive for the antiapoptotic molecule Bcl-2.33 In addition, the presence of transcripts for BSAP and Blimp-1 B cell specific transcription factors34–37 was investigated in highly purified tonsil PC, BMPC, and LPPC by RT-PCR (fig 4).

Differentiation and survival molecule expression by lamina propria (LP) and bone marrow (BM) plasma cells (PC). (A) Representative example (top series of histograms) of LPPC and BMPC (CD38h cells) expression of several differentiation molecules (CD19, CD20, HLA-DR, and VS38c) and survival associated molecules (CD95, Bcl-2). Negative control histograms are shown in grey. (B) Mean (SEM) of several experiments (n≥5) for each marker, expressed as a percentage of positive PC. *p<0.05.
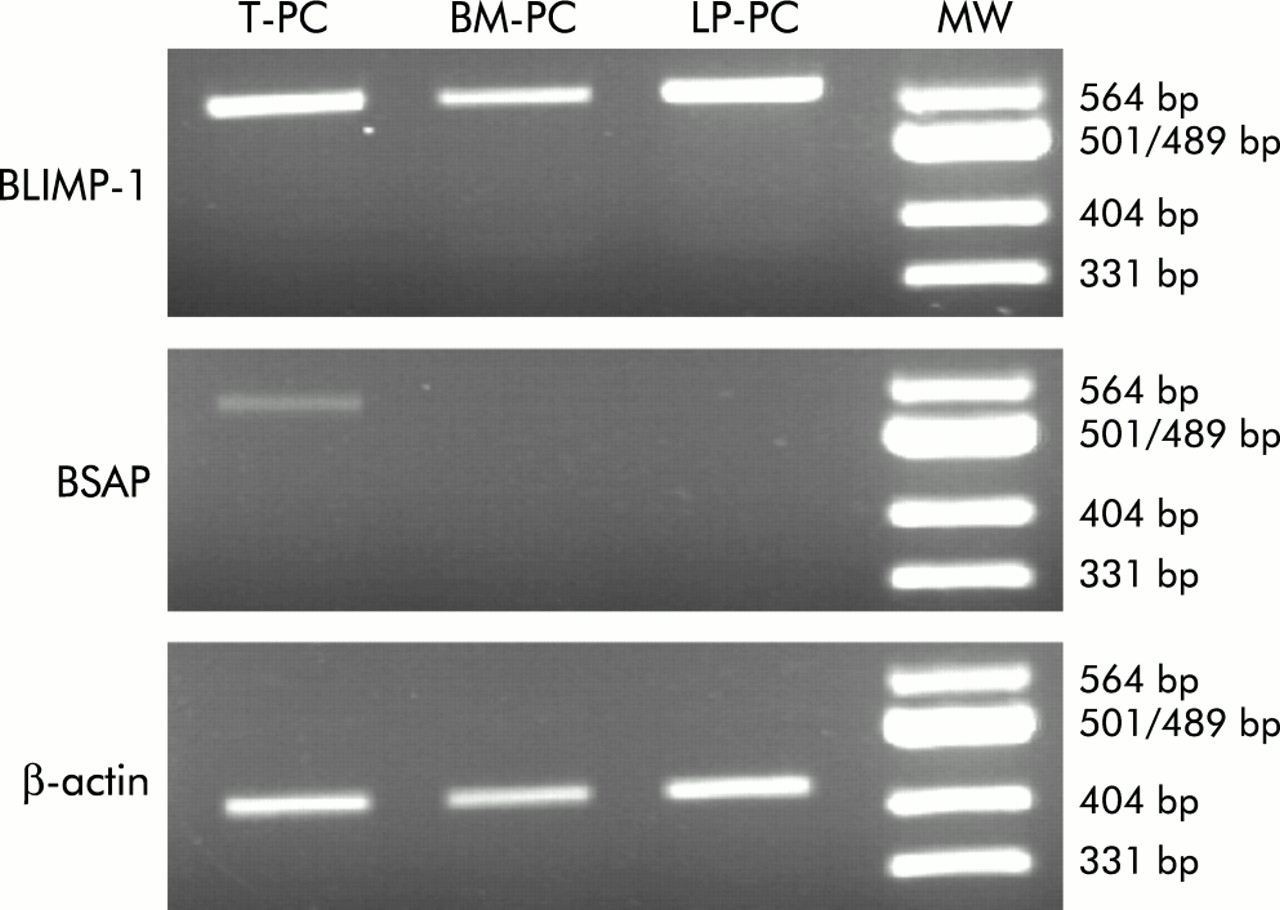
Messenger RNA for PRDI-BF1/B lymphocyte induced maturation protein 1 (Blimp 1) and B cell specific activation protein (BSAP) expression levels in different plasma cell (PC) populations. Relative amounts of Blimp-1 and BSAP mRNA were determined by reverse transcriptase-polymerase chain reaction (RT-PCR) from highly purified tonsil PC (T-PC), lamina propria PC (LP-PC), and bone marrow PC (BM-PC). RT-PCR using β-actin mRNA sequences served as an internal standard. As a molecular weight marker, a Msp I digestion of pUC19 was used (MW). Data are representative of the results of three experiments using different donors.
Comparative analysis of adhesion molecule expression by LPPC and BMPC
It is now clear that the profile of adhesion molecules expressed by a given cell plays an important role in determining its entry and attachment to specific microenvironments. Accordingly, BMPC and LPPC expression of several adhesion molecules (CD21, CD44, α4β7, CD31, CD49d, and CD54) was also evaluated using labelled mAb and flow cytometry analysis. Figure 5A shows a representative example of this study. The results obtained in all experiments are summarised in fig 5, and are represented as a percentage as well as the MFI of positive PC for each molecule under analysis (fig 5B, 5C, respectively). Although most of these markers were positive in both PC populations, the intensity of their expression differed greatly. Thus CD21 and CD44 staining was twice as high on PC from LP compared with PC from BM. α4β7 was positive on LPPC, as previously reported,38 but totally negative on BMPC. In contrast, CD31, CD49d, and CD54 staining was more pronounced on BMPC than on LPPC. The difference was specially notable for CD31 and CD49d (20-fold and 10-fold increase, respectively).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Adhesion molecule expression by lamina propria (LP) and bone marrow (BM) plasma cells (PC). (A) Representative example (upper series of histograms) of LPPC and BMPC (CD38h cells) expression of several adhesion molecules (CD21, CD44, α4β7, CD31, CD49d, and CD54). Negative control histograms are shown in grey. (B) Mean (SEM) of several experiments (n≥5) for each marker expressed as a percentage as well as mean fluorescence intensity (MFI) (C) of positive PC. *p<0.05.
DISCUSSION
Current methods of processing colon tissue to obtain LP cells generally consist of several washing steps and an overnight collagenase incubation to release lymphoid cells from LP. This procedure is time consuming and may cause damage to the functional capacities of LPPC. Accordingly, an initial aim of this study was to design a method for isolating LPPC that preserves cell function. The mechanical separation of a thin layer that only included the mucosal epithelium and the LP enabled the collagenase digestion period to be reduced to 15 minutes, which was sufficient for release of most LP cells. From this LP cell fraction, CD38h cells were highly purified by positive immunomagnetic selection through CD54, a marker that is distinctively expressed by LP CD38h cells. The correspondence between LP CD38h cells and PC was directly demonstrated by both morphology and bright intracytoplasmic anti-IgA staining criteria. Therefore, this present procedure is efficient for obtaining highly purified human colonic LPPC.
Isolated LPPC spontaneously produced large quantities of IgA in vitro. Lower amounts of IgM were also secreted into these supernatants and, in some experiments, marginal quantities of IgG secretion could be detected (data not shown). On average, IgA secretion accounted for 80% or more of the total Ig produced in vitro, a finding that is in agreement with the isotype distribution described for colon Ig secreting cells by immunohistochemical techniques.28 LPPC spontaneously produced IgA in vitro in an active way, and did so linearly for at least two weeks. This prolonged kinetics of IgA secretion clearly distinguishes LPPC from PC occurring in inductive territories such as the tonsils, lymph node, and blood,13–16 and assimilates them with BMPC, which also show prolonged kinetics of Ig secretion in vitro.20,21 On a per cell basis, the LPPC IgA secretion rate can be estimated as approximately 107 molecules per hour, which is similar to that calculated for PC by other authors.17,20 Thus LPPC isolated by this method retain their proper functional capacities.
The comparative phenotypic study of LPPC and BM PC revealed the existence of additional similarities between these cells regarding expression of a variety of differentiation markers. Both populations were CD19+/− CD20− and HLA-DRlow, and stained brightly for VS38c, denoting a well developed endoplasmic reticulum, as expected from their capacity for secreting Ig at a high rate. It is interesting to remark that the pattern of differentiation molecules shared by BMPC and LPPC markedly differs from that observed on human PC obtained in inductive areas such as the tonsils, as these latter are CD20+ CD19+ HLA-DRhigh, and show lower levels of VS38c expression,23 a finding that emphasises their relative functional immaturity.
Analysis of CD95 expression showed that an average of 50% of LPPC and 30% of BMPC carried the death receptor. Despite this fact, CD95 ligation in both PC populations had no effect on Ig secretion in culture (Medina and colleagues18 and the present study), indicating that they were not susceptible to apoptosis by this signalling mechanism. It is well established that mere expression of CD95 does not confer susceptibility to apoptosis on CD95 signalling.39 Accordingly, failure in inducing apoptosis through CD95 ligation as well as partial CD95 expression exhibited by these PC may be explained as either a residual temporary presence of this death receptor or as an inefficient or blocked death inducing signalling complex. In addition, LPPC and BMPC exhibited a high level of expression of Bcl-2, a well known antiapoptotic protein.33 These findings are clearly in contrast with those reported for human tonsils and blood PC that show higher levels of CD95, undergo apoptosis on CD95 cross linking, and contain lower amounts of Bcl-2.23 Thus low expression of CD95 and resistance to apoptosis via this receptor, together with high expression of Bcl-2, may be associated with the prolonged kinetics of Ig secretion in vitro shown by PC present in effector sites of the humoral immune response, such as BM and colon LP.
BSAP, the product of the pax 5 gene, is a B cell specific transcription factor that plays an essential role in B cell lineage commitment34 and development.40 In fact, BSAP expression is only lost at the PC stage.41 In contrast, the transcription factor Blimp-1 is expressed only in later B cell differentiation stages35 where this factor appears to control a variety of aspects of the maturation programme of PC.35 Enforced expression of BSAP inhibits the final differentiation of B lymphocytes into PC.42 It has been shown previously that the transition to terminally differentiated PC in humans is associated with the presence of the transcription factor Blimp-1 and loss of the transcription factor BSAP.23 Our results reveal that LPPC as well as BMPC expressed such a profile of transcription factors. In contrast, tonsil PC continue to express BSAP, suggesting again that PC from inductive areas appear to be less mature. Taken together, all of the results reported here establish a clear distinction between BMPC and LPPC on the one hand and PC present in inductive organs on the other, in that the latter PC appear less differentiated and exhibit a propensity to undergo apoptosis whereas the former PC show higher differentiation, survival, and functional capacity. The mechanisms involved in producing these differences remain to be elucidated.
In spite of the similarities demonstrated for human BMPC and LPPC, analysis of their respective profiles of adhesion molecule expression reveals marked differences. Thus CD21 and CD44 expression was brighter on LPPC than on BMPC. Moreover, expression of the integrin α4β7 was only found on LPPC. In contrast, CD31, CD49d, and CD54 showed a higher presence on BMPC than on LPPC. Taken together, these findings demonstrate the occurrence of specific patterns of adhesion molecule expression for each PC population, probably related to the ligands occurring in their particular microenvironment. High expression of CD49d and CD31 by BMPC probably indicates a role for these two molecules in the homing and attachment of PC to BM specific niches. The integrin α4β7 plays a key role in the homing of lymphoid cells to mucosal tissues including the LP, as has been demonstrated in in vivo animal models43–45 and, more indirectly, in human in vitro adhesion assays.46 Little is known of the specific adhesion molecules used by circulating PC precursors to migrate into the LP. In this respect, B cell blasts detected in microlymphatic vessels of Peyer’s patches are α4β7+,47 and Ab secreting cells released to the circulation after oral vaccination are exclusively detected in the α4β7+ cell fraction.48 These observations, together with the finding that LPPC are also α4β7+, appear to indicate that this integrin might act in the recognition of mucosal endothelium by LPPC precursors. Expression of α4β7 by LPPC was weak in comparison with that of CD21, CD44, CD49d, or CD54. Therefore, adhesion between PC and the LP microenvironment could depend on some of these molecules, or perhaps others not tested, rather than on α4β7. Further work will be required to ascertain these possibilities.
In summary, LPPC and BMPC have apparently reached the same terminal differentiation stage within the PC compartment, as can be deduced from their identical Ig secreting and survival capacity, and their common expression of differentiation markers. These characteristics seem to define PC occurring in effector areas of the humoral immune response (BM and LP), and distinguish them from those PC present in inductive territories. Nevertheless, BMPC and LPPC differ markedly in their adhesion molecules, in relation to their different localisation requirements. This study describing the phenotype and functional capabilities of normal PC from human intestinal LP could be helpful in the analysis of PC in intestinal diseases with an altered humoral immune response.
Acknowledgments
We thank Dr J Martorell and Dr J Pérez-Requena for help in the histological studies. The study was supported by grant 01/1590 from Fondo de Investigaciones Sanitarias, Spain.