Article Text

Abstract
We observed the development of phenotypic hereditary haemochromatosis in a non-hereditary haemochromatosis liver transplant recipient, following transplantation with a liver from a C282Y heterozygous donor. No cause for secondary iron overload was identified. Subsequent sequencing of the HFE gene of both donor and recipient revealed a strong candidate for a novel pathogenic HFE mutation. In the recipient, heterozygosity for a single base substitution in exon 1, g.18 G>C, resulting in the substitution of arginine by serine at codon 6 (R6S), was detected. This R6S variation is likely to represent a novel pathogenic missense mutation of the HFE gene. An interaction between R6S heterozygosity in the recipient and C282Y heterozygosity in the donor liver is the most likely explanation for the development of iron overload in this patient. The report suggests that an hepatic defect is required for expression of hereditary haemochromatosis and that the intestinal HFE genotype is not the exclusive determinant of iron status. It also raises the possibility that a hereditary haemochromatosis phenotype may result from transplantation of C282Y heterozygous donor livers into recipients with heterozygous pathogenic HFE mutations. This possibility may have significant implications for the common practice of transplanting C282Y heterozygous livers.
- HH, hereditary haemochromatosis
- OLT, orthotopic liver transplantation
- HIC, hepatic iron concentration
- HII, hepatic iron index
Statistics from Altmetric.com
- HH, hereditary haemochromatosis
- OLT, orthotopic liver transplantation
- HIC, hepatic iron concentration
- HII, hepatic iron index
Transplantation of C282Y heterozygous livers is considered to be a safe practice and such livers are frequently transplanted (up to 17% of donors in some centres).1 Current opinion, which favours the intestine as the exclusive site of the defect in hereditary haemochromatosis (HH) and the requirement for homozygous HFE mutations at this site, has provided reassurance that iron overload should not occur. In addition, the only study to investigate the safety of C282Y heterozygous liver transplantation did not find a survival difference between patients transplanted with C282Y heterozygous livers and patients transplanted with normal livers.2 However, in this report we provide the first description of a HH phenotype developing in a non-HH recipient following transplantation of a C282Y heterozygous donor liver. The subsequent discovery of a novel heterozygous mutation of the recipient’s HFE gene is described and we speculate on the mechanisms involved in the development of the HH phenotype. The contribution of this case to our understanding of the role of the liver genotype in HH expression and the clinical implications of this case are also discussed.
CASE REPORT
A 54 year old Caucasian male with alcoholic cirrhosis (Child-Pugh class C) was referred for orthotopic liver transplantation (OLT) assessment. The patient had maintained abstinence from alcohol during the previous six months and had no other significant past medical history or family history of iron overload or liver disease.
There was no evidence of HH in the recipient prior to OLT. The common HFE mutations (C282Y and H63D) were absent. Pre-transplant liver biopsy demonstrated inactive micronodular cirrhosis with minor grade 1 siderosis (less than 25% of hepatocytes affected) with small amounts of stainable iron seen in occasional hepatocytes and portal tract macrophages, in keeping with an alcoholic aetiology. The hepatic iron concentration (HIC) was 30 μmol/g dry weight (normal <40) and the hepatic iron index (HII) was 0.7 (HH>1.9).
The 44 year old Caucasian female donor was heterozygous for the C282Y mutation. Biopsy of the donor liver taken at the time of transplantation demonstrated only mild iron stores consistent with C282Y heterozygosity (grade 1 siderosis, HIC=41 μmol/g, HII=0.9).
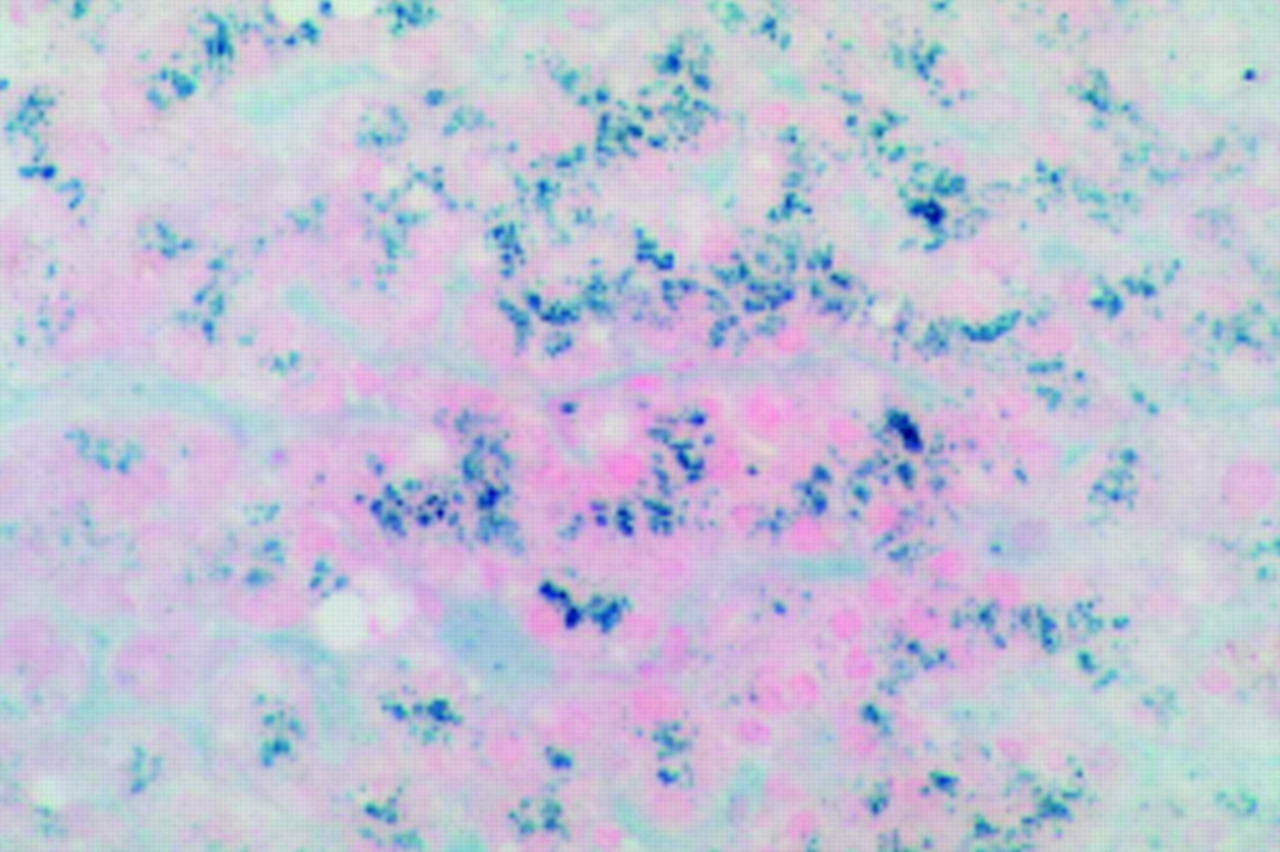
The postoperative course and first four years after transplantation were uneventful. During repair of an incisional hernia, 49 months following OLT, a further liver biopsy was taken (fig 1). Surprisingly, the major feature of this biopsy was prominent iron deposition within hepatocytes, amounting to grade 4 siderosis (>75% of hepatocytes affected). HIC was 252 μmol/g dry weight and HII 5.0. Repeat liver biopsy again demonstrated grade 4 siderosis and similar values for HIC and HII. Subsequent venesection removed greater than 6 g of iron prior to the onset of iron deficiency, which had accumulated in only four years.

Perls’ stain of a donor liver biopsy section taken at 49 months. Significant parenchymal iron deposition (grade 4 siderosis) is present, consistent with a hereditary haemochromatosis phenotype.
A thorough search for causes of acquired iron overload did not reveal any risk factors. The patient had not been excessively transfused and did not take iron supplements. There was no suggestion of alcohol recidivism on post transplant liver biopsies or on frequent historical enquiry. The transplanted liver was not cirrhotic and no significant rejection episodes were recorded. There was no evidence of hepatitis C, non-alcoholic steatohepatitis, or significant portosystemic shunting which have all been associated with siderosis. Acquired iron overload from a chronic haemolytic or iron loading anaemia was unlikely given normal blood film, haptoglobin, direct antiglobulin test, lactate dehydrogenase, and unconjugated bilirubin levels. In addition, iron deposition was predominantly parenchymal, a pattern suggestive of HH rather than acquired iron overload.
To help explain the development of HH in this case, sequencing the HFE gene of both donor and recipient was performed. Genomic DNA was extracted from peripheral blood lymphocytes and the essential coding regions and all intron-exon boundaries of the HFE gene were amplified as three fragments: fragment 1 (−108→IVS1+186); fragment 2 (IVS1–101→IVS3+70); and fragment 3 (IVS3–63→IVS6+63). Primers used were as follows:
HH1f=TCCTGAGCCTAGGCAATAGC,
HH1r=CCTATCTGCAGTTCAGTGGT,
HH2f=CCAGACACAGCTGATGGTAT,
HH2r=GATTCCGTGCCCTGCAACCT,
HH3f=TTCCAGTCTTCCTGGCAAG,
HH3r=ATGCACTCCCTCTTTGAGTC.
Polymerase chain reaction fragments were sequenced directly using the amplification primers and Applied Biosystems BigDye Terminator Cycle Sequencing Kit. The sequence of both DNA strands was determined for each fragment and compared with the reference sequence (GenBank reference sequence Z92910) using Sequence Navigator software.
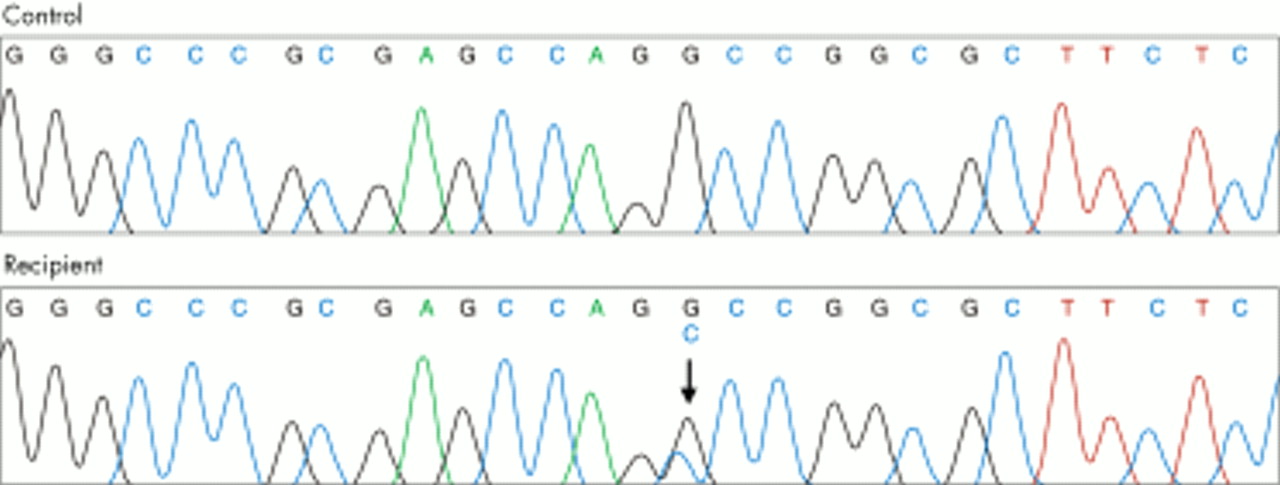
From this sequencing a strong candidate for a novel pathogenic HFE mutation emerged. The recipient was heterozygous for a single base substitution in exon 1, g.18 G→C, resulting in the substitution of arginine by serine at codon 6 (R6S). This variation has not been previously described and is demonstrated in fig 2.
{kind=link}
{kind=link}
DNA sequence electropherogram from a normal control and the liver transplant recipient. The same sequence in exon 1 is shown. The recipient is heterozygous for a G→C base substitution at nucleotide 18 (arrow).
Thirty five European subjects, with no history of HH, were tested for the presence of the codon 6 variation (R6S). The R6S variation was not detected in any of these 70 alleles. We were able to screen six of eight of the recipient’s living first degree relatives (mother, three siblings, and two children) for both phenotype (transferrin saturation and ferritin) and genotype (presence of the R6S mutation). All of these individuals demonstrated normal iron studies. The R6S mutation was detected in four of these six family members (52 year old brother, 48 year old sister, 30 year old daughter, and 28 year old son), who were all heterozygous for the mutation and did not demonstrate other common HFE mutations (C282Y, H63D).
DISCUSSION
The R6S variation discovered in this case is likely to represent a novel rare pathogenic missense mutation of the HFE gene and adds to a growing number of rare HFE mutations which have been associated with the disease.2–4 Support for its role as a pathogenic mutation comes from its association with a HH phenotype in this case and its absence from a normal control group. Further support comes from the position of this single amino acid substitution, in the important signal sequence of the HFE gene, required for transport of the nascent polypeptide across the membrane of the endoplasmic reticulum. Although pathogenic mutations have not been previously described from this region of the HFE gene, examples of pathogenic signal sequence mutations in other genes exist.5–7 Substitution of a neutral amino acid (arginine) for a hydrophilic amino acid (serine), caused by the R6S mutation, is likely to have functional consequences because signal sequences require hydrophobic amino acids for efficient translocation across the endoplasmic reticulum and protein processing.5
Despite this discovery, explanation for the development of phenotypic HH in this case remains difficult. Current opinion favours the intestine as the exclusive site of the defect in HH and the requirement of homozygous HFE mutations at this site for expression of HH. Individuals who are heterozygous for pathogenic HFE mutations rarely if ever develop significant iron overload in the absence of cofactors.8 It therefore appears unlikely that the heterozygous R6S mutation alone could explain the development of HH in this case. The possibility of the R6S mutation having an autosomal dominant pattern of expression is also unlikely as there was no family history of iron loading diseases. In addition, the four family members who were heterozygous for the R6S mutation all had normal iron studies, despite reasonably advanced ages in the case of the two affected siblings.
DNA from recipient duodenum and donor liver was also compared with the results obtained from lymphocyte DNA sequencing. Duodenal (R6S, wild-type) and liver (C282Y, wild-type) HFE genotype corresponded to recipient and donor HFE genotype, respectively. Therefore, the possible contribution to the HH phenotype of recipient chimerism or mosaicism of the liver following transplantation was excluded.
That the heterozygous C282Y donor liver mutation alone was responsible for expression of HH also seems unlikely as the mutation was heterozygous only and was not present in the recipient intestine.
Although we are unable to exclude contributory mutations in areas not covered by our sequencing, the possibility of non-HFE haemochromatosis seems remote. Juvenile haemochromatosis (HFE 2) is characterised by an earlier onset and a more severe clinical course than was seen in our patient.9 Mutations in the transferrin receptor 2 gene (HFE 3) have only been described in a small number of Italian families.10 Mutations in the ferroportin 1 gene (HFE 4) are associated with an autosomal dominant form of haemochromatosis with iron deposition in Kupffer cells and a poor tolerance to venesection therapy.11–14 Our patient did not demonstrate any of these features.
The most plausible explanation therefore for the rapid expression of phenotypic HH following OLT in this case appears to be the combination of a novel heterozygous HFE mutation in the recipient (R6S) and a heterozygous HFE mutation in the donor liver (C282Y). This conclusion has several interesting implications.
It suggests that both an intestinal and hepatic defect are required for expression of HH rather than an exclusive intestinal defect. This concept could help explain several anomalies relating to HH expression after transplantation which cannot be understood using our current framework. Firstly, HH patients do not develop iron overload following transplantation with normal livers, despite adequate follow up.15 A lack of HFE mutations in the donor liver could explain this. Secondly, iron loaded livers from HH individuals rapidly lose iron following inadvertent transplantation into non-HH recipients.16–18 This could be explained by a lack of HFE mutations in the recipient intestine. Conversely, the single case of persistent iron overload following inadvertent transplantation of an iron loaded liver into a non-HH recipient could be explained by the presence of a heterozygous HFE mutation in the recipient’s intestine.19 Significantly, these cases were described prior to the discovery of the HFE gene and the HFE status of donors and recipients was therefore not reported. Finally, iron accumulation following inadvertent transplant of a C282Y homozygous liver and intestine is also consistent with the hypothesis that combined intestinal and liver defects are required for expression of HH.20
A second interesting implication is that heterozygous HFE mutations in the intestine and liver may result in a HH phenotype following liver transplantation. If this were true we would anticipate the development of phenotypic HH following transplantation of a C282Y heterozygous liver into a C282Y heterozygous recipient. Given the frequency of this situation (approximately 1.5% of all transplants), it is surprising that this phenomenon has not been previously described. Perhaps a long lag time is required for the development of HH in this setting. Alternatively, the development of HH may be specific to the heterozygous R6S and C282Y combination. Studies identifying C282Y heterozygous recipients, transplanted with C282Y heterozygous livers, and providing long term follow up have not been done. Given the scarcity of donor organs and the high prevalence of the C282Y mutation, such studies are now required to help resolve this question and to reassure us that transplantation of C282Y heterozygous livers does not lead to iron overload in recipients with heterozygous HFE mutations.