Article Text

Abstract
Aim: Hyperplastic polyps (HP) of the colorectum have traditionally been regarded as non-neoplastic lesions. Recent data implicate HP in the pathogenesis of colorectal cancers (CRC) characterised by extensive DNA methylation and microsatellite instability. The aim of this study was to identify morphological and molecular features that may characterise subtypes of HP with potential for neoplastic progression.
Materials and methods: HP (22) clustering around distal CRC (group I) were compared with HP (58) in subjects with hyperplastic polyposis (group II). DNA methylation was tested in methylated in tumour (MINT) loci (1, 2, 12, 31) and genes HPP1, MGMT, p14ARF, p16INK4a, and hMLH1.
Results: Group II HP showed significantly more methylation than group I HP at all loci except MINT1 and MGMT. Group I showed the lowest frequency of DNA methylation but the highest frequency of K-ras mutation. Group II HP (termed HP variant) had the morphological features of the recently described “sessile serrated adenomas”. Methylation of hMLH1 was found most frequently in group II polyps that included foci of dysplasia (7/10) and in no group I lesions.
Conclusion: The findings support the existence of morphological and molecular heterogeneity among HP and highlight a subset that is likely to have significant malignant potential.
- hyperplastic polyposis
- CpG methylation
- colon cancer
- microsatellite instability
- ACF, aberrant crypt foci
- AD, adenoma
- CIMP, CpG island methylator phenotype
- COBRA, combined bisulphite restriction analysis
- CRC, colorectal cancer
- HP, hyperplastic polyps
- HP variant, hyperplastic polyp variant
- MGMT, O-6-methylguanine DNA methyltransferase
- MINT, methylated in tumour
- MP, mixed polyps
- MSI, microsatellite instability
- MSI-H, high level MSI
- MSI-L, low level MSI
- MSP, methylation specific polymerase chain reaction
- MSS, microsatellite stable
- PCR, polymerase chain reaction
- SA, serrated adenomas
- VA, villous adenoma
Statistics from Altmetric.com
- ACF, aberrant crypt foci
- AD, adenoma
- CIMP, CpG island methylator phenotype
- COBRA, combined bisulphite restriction analysis
- CRC, colorectal cancer
- HP, hyperplastic polyps
- HP variant, hyperplastic polyp variant
- MGMT, O-6-methylguanine DNA methyltransferase
- MINT, methylated in tumour
- MP, mixed polyps
- MSI, microsatellite instability
- MSI-H, high level MSI
- MSI-L, low level MSI
- MSP, methylation specific polymerase chain reaction
- MSS, microsatellite stable
- PCR, polymerase chain reaction
- SA, serrated adenomas
- VA, villous adenoma
There is evidence that colorectal cancer (CRC) does not evolve through a single sequence of molecular and morphological alterations but by different pathways with their own clinical, pathological, and molecular characteristics.1 The two most well documented of these are the chromosomal instability and microsatellite instability (MSI) pathways.2,3 A particular precancerous sequence implicating lesions with a serrated crypt architecture has been linked with the MSI pathway. Epithelial lesions with glandular serration encompass aberrant crypt foci (ACF), hyperplastic polyps (HP), mixed polyps (MP), and serrated adenomas (SA).4
The existence of MP provides evidence for a transition from HP to adenoma (AD) or SA.5 However, compared with MP and SA, both ACF and HP are common lesions and the great majority will not transform into clinically significant polyps. The existence of heterogeneity with respect to clinical behaviour is matched by good evidence for molecular heterogeneity among serrated lesions. In particular, the K-ras mutation has been detected with high frequency in ACF6,7 whereas the frequency of this alteration in HP,8 SA,9,10 and sporadic CRC with high level MSI (MSI-H) decreases progressively.11 On the other hand, a high frequency of DNA methylation is described in SA12 and sporadic MSI-H CRC.13 This alteration occurs in HP although less frequently.14
Lesions that show extensive DNA methylation have been described as having the CpG island methylator phenotype (CIMP).15 This alteration has been associated with the process of aging as well as neoplasia.16 An important effect of DNA methylation is silencing of the promoter region and therefore inactivation of the gene. There is a link between methylation and MSI, with methylation of the DNA repair gene hMLH1 being the principal mechanism underlying the pathogenesis of sporadic MSI-H CRC.17 Methylation of another DNA repair gene, O-6-methylguanine DNA methyltransferase (MGMT), has been linked with low level MSI (MSI-L).18 Among the additional genes that may be methylated in colorectal neoplasms are HPP1, p14ARF, and p16INK4a that are implicated in the control of cell adhesion, apoptosis, and cell cycle control respectively.15,19,20 Anonymous CpG sequences known as methylated in tumours (MINTs) have been used to determine CIMP status. Among these, MINTs 1, 2, 12, and 31 have been found to be particularly sensitive and reliable.13,21,22
A specific clinical situation in which HP may show concordant and extensive DNA methylation is hyperplastic polyposis.14 Hyperplastic polyposis is increasingly being associated with CRC that presents at a relatively early age and in which MSI is overrepresented.23–26 The polyps presenting in hyperplastic polyposis have been distinguished on morphological grounds from the common small HP that typically present in the distal colon and rectum.27 The distinguishing features encompass size, architecture, differentiation, and proliferation. Similar appearing HP-like polyps may also occur sporadically.28,29 Unlike typical HP, these variants frequently present within the proximal colon. These have been named “sessile serrated adenomas” to distinguish them from the more frequent HP that remain small and are localised to the distal colon and rectum.30
The aim of this study was to undertake a comprehensive morphological and molecular characterisation of serrated lesions (ACF, HP, MP, and SA) presenting in two contrasting clinical scenarios. The first scenario is distal CRC in which HP cluster as “satellite” lesions around the tumour. The second scenario is hyperplastic polyposis with or without coexisting CRC. The underlying hypothesis was that within well defined clinical scenarios it might be possible to assemble particular classes of serrated lesions into a linear morphogenetic sequence culminating in CRC. By contrast, other serrated lesions representing the majority may be grouped into a non-progressing or benign category.
MATERIALS AND METHODS
Patients and specimens
The study was performed on freshly obtained lesions identified in 12 surgical specimens of colorectum that were resected at the Royal Brisbane, Princess Alexandra and Wesley Hospitals in Brisbane, Queensland, between 2000 and 2002. Matched normal colorectal mucosa was obtained from the resection margin that was furthest from any malignant lesion. None of the patients had preoperative chemotherapy or radiotherapy except for patient I.2 where the cancer specimen was not analysed. In nine cases, additional samples of DNA were subsequently obtained from formalin fixed specimens. Ethics approval was obtained and all subjects provided informed written consent. One half of the polyp was immediately snap frozen while the other half was submitted for routine diagnostic assessment.
Based on review of the final pathology report and additional information from the clinical records, but without knowledge of the molecular findings, patients were classified into two groups according to the number, size, and anatomical location of HP. Group I patients had one or more small HP that clustered around a cancer of the distal colon or rectum in a satellite-like distribution.30 Group II patients satisfied one or more of the following criteria for hyperplastic polyposis: (1) at least five HP proximal to the sigmoid colon with two being greater than 10 mm in diameter; (2) any number of HP and a family history of hyperplastic polyposis; and (3) at least 30 HP in a pancolonic distribution (table 1).31
Clinical characteristics of patients with hyperplastic polyps
Histological classification of polyps
Lesions were initially classified as ACF, HP, MP, and SA. Polyps were subsequently reviewed by two independent observers with the aim of distinguishing classical HP and a HP variant that has been described as “sessile serrated adenoma”.27,28 In this study, the latter are referred to as a “HP variant” as they do not show features of adenomatous dysplasia (fig 1). Features used to discriminate the HP variant from traditional HP included: (1) exaggerated serration, papillarity, or villosity including serration in the lower crypt; (2) crypt irregularity, including pronounced budding, branching, or horizontally arranged crypts; (3) crypt dilatation; (4) increased crypt epithelium to stroma ratio; (5) mitoses in the upper half of crypts; (6) vesicular nuclei with prominent nucleoli in the upper half of crypts; and (7) increased production of intracellular and/or luminal mucin.28,29 A polyp was designated as a HP variant when four or more of these features were present. A polyp was designated MP if the HP variant contained obvious foci of SA and/or tubular adenoma. For the review process, polyps were relabelled and coded anonymously so that the observers could not know the group of origin. In the case of disagreements a final diagnosis was reached by consensus.
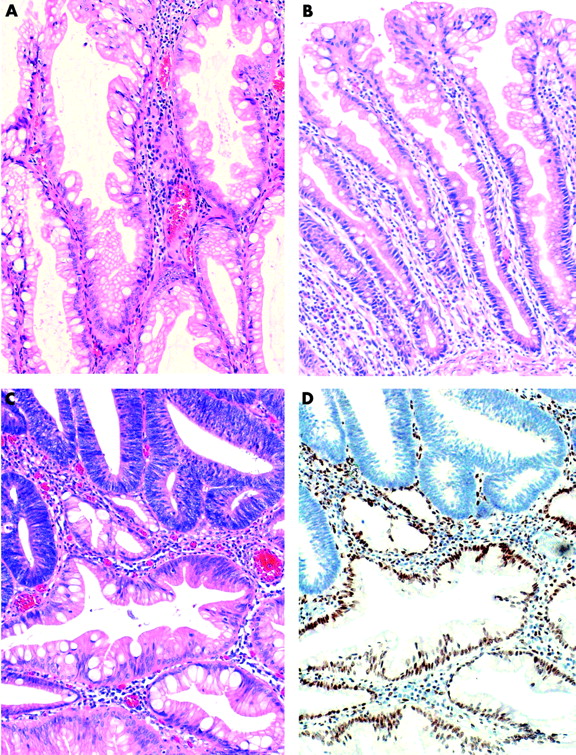
(A) Hyperplastic polyp variant (HP variant) from a group II patient showing dilated crypts and exaggerated serration but no evidence of adenomatous dysplasia (haematoxylin-eosin). (B) Typical hyperplastic polyp (HP) from a group I patient in which crypt architecture is minimally altered and serration is mainly confined to the upper crypt epithelium. (C) Mixed polyp, partly HP variant and partly adenomatous. (D) Adenomatous glands show loss of expression of hMLH1.
Immunohistochemistry for hMLH1 and MGMT staining protocols have been described in detail elsewhere.18,23
DNA extraction, MSI, and K-ras analysis
Where fresh tissue was available, DNA was extracted using a NucleoSpin DNA extraction kit (Macherey-Nagel, Duren, Germany) or the Chelex method from formalin fixed tissue.32 DNA was examined for MSI using 6–10 microsatellite markers.33 MSI-H was defined as more than 40% of markers showing instability and MSI-L as fewer than 40%. Mutations in codons 12 and 13 of the K-ras gene were identified by the polymerase chain reaction (PCR)-restriction fragment length polymorphism technique using restriction enzymes BstN1 and Hph1.34
Methylation detection by COBRA and MSP
Methylation of the 5′ CpG promoter region of three genes, p14ARF,35p16INK4A,36 and MGMT,37 was determined by methylation specific PCR (MSP). These three genes have been employed in several earlier methylation studies of CIMP and colorectal cancer.
Combined bisulphite restriction analysis (COBRA) for hMLH1 and HPP1 methylation was performed as described previously38 with inclusion of nested primers for hMLH1 for increased sensitivity (table 2). The COBRA assay for hMLH1 measured the methylation state of the CpG at position −75 from the start codon ATG. Genomic DNA was always used as a negative control for MSP PCRs. Blood DNA treated in vitro with SssI methyltransferase was used as a positive control for methylated alleles.
List of primers for determination of the methylation status of methylated in tumours (MINTs) and genes by combined bisulphite restriction analysis and methylation specific polymerase chain reaction
MINT1, MINT2, MINT12, and MINT31 were assayed by COBRA (table 2). For DNA extracted from formalin fixed tissue, the MSP method for MINTs described by Chan and colleagues14 was used. Because tumours contained a variable amount of non-cancer tissue, no attempt was made to quantitate levels of methylation, which were scored as either positive or negative (fig 2).

Polymerase chain reaction (PCR) products showing the methylation status of methylated in tumours (MINTs) and genes, and mutation status of K-ras. (A) (i) Methylation of MINT1, (ii) MINT2, (iii) MINT12, and (iv) MINT31. When methylated, new PCR products appear after incubation with restriction enzymes. (B) (i) Methylation of hMLH1, (ii) HPP1, (iii) MGMT, (iv) p14ARF, (v) p16INK4A, (vi) K-ras codon 12, the mutant product is larger than the wild-type, and (vii) K-ras codon 13, the mutant product is smaller than the wild-type. MGMT, p14ARF, and p16INK4A were assayed by methylation specific PCR where the appearance of a PCR product indicates methylation. N, normal colon mucosa; Ca, cancer; P, polyp.
Criteria for CIMP+
Lesions were classified as CIMP-neg, CIMP-low, and CIMP-high if none, 1–2, or 3–4 MINTs were methylated, respectively.
Statistical analysis
Statistical probabilities were analysed by 2×2 contingency tables using binomial distribution of differences and either Fisher’s exact test or Pearson’s χ2 test where appropriate with a StatXact package (Cytel Corporation, Chicago, Illinois, USA). Probability (p) values of ⩽0.05 were considered significant.
RESULTS
Classification of polyps
Details of the number and anatomical distribution of HP are provided in table 1. Traditional adenomas were uncommon and are not included in the overall analysis. Following review, only two of the 22 group I HP were reclassified, giving a total of 20 traditional HP, one HP variant, one SA, two villous adenomas, and six CRC. By contrast, 19 of 24 available group II polyps that were originally classified as traditional HP were reclassified as HP variant. As the distinction between HP and HP variant is still controversial and reclassification resulted in low numbers of HP variant and HP in groups I and II, respectively, the analyses do not distinguish HP variant from HP unless stated otherwise. Group II lesions totalled 47 HP, nine MP, one SA, one AD, and four CRC samples. All nine MP were combinations of HP variant and adenoma (fig 1C), the latter being serrated in two cases.39
Methylation in polyps and cancers
MINTs were methylated differently in group I and II polyps whether this was based on individual MINTs or the total number of MINTs (table 3). Of a total of 58 polyps in group II, 35 were CIMP-high, 16 were CIMP-low, and seven were CIMP-neg. Of 24 group I polyps, three were CIMP-high, 11 were CIMP-low, and 10 were CIMP-neg (p<0.003 value calculated for CIMP-high and CIMP-low v CIMP-neg in group I compared with group II). Two of the three CIMP-high polyps in group I included the single HP variant and the single SA from this group. Both of these occurred in patient I.7 with a CIMP-high CRC.
Methylation of four methylated in tumours (MINTs 1, 2, 12, and 31) and five genes (hMLH1, HPP1, MGMT, p14ARF, p16INK4A) of polyps in satellite hyperplastic polyps (HP) associated with distal cancers and hyperplastic polyposis. The mutation rate in K-ras (codon 12 or 13) is also indicated
Methylation of hMLH1 was restricted to group II polyps. Within this subset, hMLH1 methylation was found in 13 of 58 polyps. However, this included seven of 10 (60%) MP with foci of dysplasia (table 3). The non-dysplastic polyps with hMLH1 methylation had all been independently reclassified as HP variant. All but two of the 13 polyps with hMLH1 methylation were CIMP-high, the other two being CIMP-low. It is also notable that the one AD in patient II.2 among nine HP variants showed no methylation of MINTs or genes (fig 3). Two of the three polyps with features of SA in group II showed methylation of hMLH1.
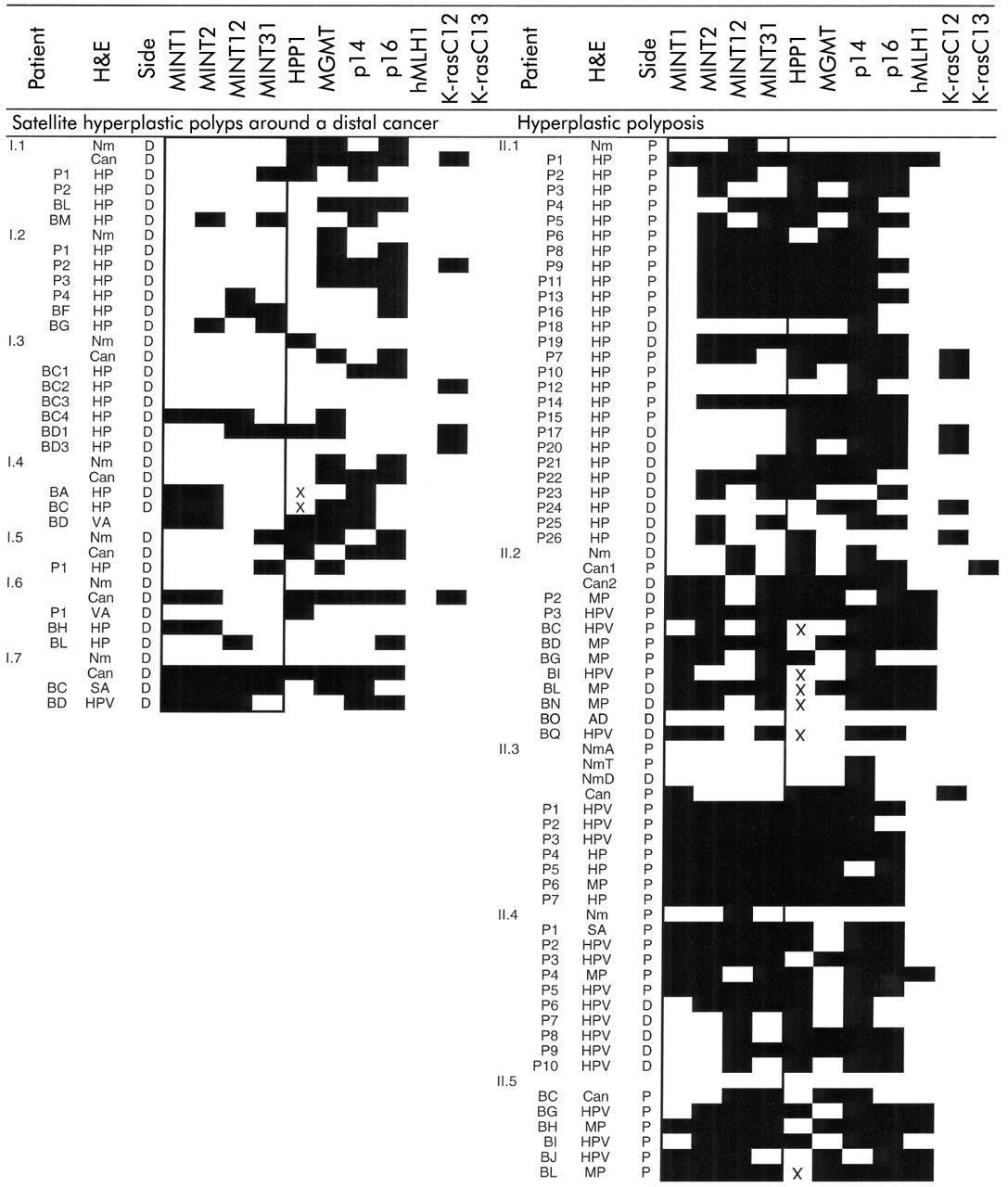
Methylation pattern of methylated in tumours (MINTs) and genes in polyps, cancers, and normal colon mucosa. The shaded area indicates methylation of a MINT or gene; a blank indicates no methylation. The box highlights the methylation of MINTs. A mutation in codon 12 or 13 of K-ras is indicated by a shaded area. An X indicates no polymerase chain reaction product was obtained. Group I patients (Nos I.1–I.7) had one or more hyperplastic polyps (HP) in a satellite distribution around a distal cancer. Group II patients (Nos II.1–II.5) fulfilled the criteria of hyperplastic polyposis. P, fresh polyp tissue available for DNA analysis; B, archival material. The haematoxylin-eosin (H&E) stained lesions are identified as HP, HP variant (HPV), serrated adenoma (SA), mixed polyp (MP), adenoma (AD), villous adenoma (VA), cancer (Can), and normal mucosa (Nm). Ascending, transverse, and descending normal mucosa is indicated by NmA, NmT, and NmD, respectively. The side of the colon the specimen was located is indicated as proximal (P) or distal (D). There was no normal tissue available for patient II.5.
Two of the HP variants from patient II.2 had foci of high grade dysplasia amounting to carcinoma in situ, and were classified as cancer for the purposes of this study. One of the samples was CIMP-high with additional methylation of HPP1, MGMT, p14ARF, and p16INK4a, and the second was CIMP-low with additional methylation of HPP1, p14ARF, p16INK4a, and a K-ras mutation (fig 3). Neither showed methylation of hMLH1 despite the high frequency of hMLH1 methylation in multiple lesions with less severe atypia from the same subject. The cancer specimen from patient II.5 was CIMP-low with additional methylation of MGMT and p14ARF and without methylation of hMLH1, again despite the finding of multiple polyps with hMLH1 methylation in the same subject. The fourth cancer from a subject with hyperplastic polyposis (II.3) was CIMP-low with additional methylation of HPP1, MGMT, and p14ARF and mutation of K-ras. Of the cancer samples from group I patients, four were CIMP-neg, one was CIMP-low, and one was CIMP-high. The four CIMP-neg cancers all showed methylation of at least two genes. As noted above, patient I.7 with a CIMP-high cancer also had a CIMP-high HP variant (the only HP variant diagnosed in group I) and a CIMP-high SA.
K-ras mutation
K-ras mutation was detected in 10 of 82 (12.2%) polyps from across both groups, and four of 11 (37%) CRC samples (table 3). None of the 14 K-ras mutations occurred in lesions that were MINT-high or had methylation of either MINT1 or hMLH1. However, eight lesions with K-ras mutations were MINT-low and genes other than hMLH1 were frequently methylated in samples with K-ras mutations. All K-ras mutations in group II HP were restricted to a single subject with hyperplastic polyposis (II.1). In this subject, HP were less methylated compared with the other subjects with hyperplastic polyposis. MINT1 was not methylated in any of these polyps.
Size, location, and methylation of HP
The 26 HP from patient II.1 with hyperplastic polyposis were stratified according to size (<5 or 6–15 mm) to determine if they were related to the extent of MINT methylation. There was a significant correlation between size and CIMP-high methylation, with the larger hyperplastic polyps being more extensively methylated (p<0.005). The location of HP in all subjects within the proximal (up to the splenic flexure) versus the distal colon was compared with their MINT status to determine if methylation of MINTs was associated with proximal location in the large colon. Although most of the HP in hyperplastic polyposis patients were obtained from the proximal colon, MINT-high status was demonstrated in seven of 20 (35%) distal polyps whereas the same was found in only three of 24 (12.5%) distal group I HP (p = 0.038, one-sided).
DISCUSSION
We found that traditional HP from group I patients had very little methylation of MINTs or genes. In contrast, group II patients, clinically fulfilling the criteria of hyperplastic polyposis, were heavily methylated both at MINTs 2, 12, and 31 and for genes, HPP1, p14ARF, and p16INK4a. In particular, MINTs were very discriminating between the two groups. When MP were included in group II HP, MINT1 methylation was significantly different from group I HP lesions. This could indicate that MINT1 is methylated at a later stage than other MINTs in HP in hyperplastic polyposis.
The decision to collect lesions presenting in two contrasting clinical scenarios served to highlight the molecular differences across the various classes of lesion and excludes a single linear model of accumulating genetic alterations. Serrated lesions and polyps of the colorectum, encompassing ACF, HP, MP, and SA, are unlikely to represent different stages of a single pathway of morphogenesis. ACF39 and HP are common lesions compared with MP and SA,4 indicating that the majority do not progress along the “serrated pathway”.
Classification of “serrated polyps” has become complex and somewhat controversial in recent years and cannot be fully resolved in the present study. When SA were first highlighted it was appreciated that some resembled HP whereas others had a more adenomatous appearance.4 Lesions termed HP variant in this study were conceived as occupying the HP end of the serrated spectrum yet did show features that have been associated with adenomas. On this basis and in order to address the possibility that these lesions are precancerous, others have introduced the term “sessile serrated adenoma”.27–29 The recent recognition that serrated polyps share two molecular alterations, namely CIMP-high status and mutation of BRAF, with sporadic MSI-H CRC, strengthens the concept of a serrated pathway of colorectal tumorigenesis.40–42
The flow diagram illustrates our concept of the early stages of the serrated neoplasia pathway leading to MSI-H CRC (fig 4). Serrated lesions with K-ras mutations include ACF and traditional HP but these have little or no potential for neoplastic evolution. There are two pathways to an SA or MP, both implicating CIMP-high status, mutation of BRAF, and loss of function of the DNA repair genes MGMT and hMLH1 that are inactivated by promoter hypermethylation. There may be another pathway via HP variants that are CIMP-high but have a K-ras mutation instead of BRAF, and MGMT inactivated rather than hMLH1. We have published evidence for this group of MSI-L/MSS CRCs.22

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Aberrant crypt foci (ACF) and hyperplastic polyps (HP) are conceived as non-progressing lesions despite the frequent finding of K-ras mutations. The genetic alterations underlying the HP variant implicate the same signalling pathway (BRAF being downstream of RAS) and a greater extent of DNA methylation. The potential for malignancy of the HP variant is more likely to be realised when key DNA repair genes hMLH1 or MGMT are methylated and inactivated. Subsequent mutations will result in the development of dysplasia giving a serrated adenoma (SA) or mixed polyp (MP) and finally colorectal cancer (CRC). The possibility that some HP variants arise through synergy between K-ras and the CpG island methylator phenotype (CIMP) is included in the model but probably applies more to microsatellite stable (MSS)/low level microsatellite instability (MSI-L) CRC than high level MSI (MSI-H) CRC in which the BRAF mutation dominates.
There were no differences in the frequency of K-ras mutations across groups I and II. However, all K-ras mutations in group II polyps were restricted to a single subject (II.1). This patient differed from other subjects with hyperplastic polyposis in multiple respects. She did not have cancer and HP were more numerous and showed less methylation of MINTs (although more methylation than in HP from group I subjects). Lack of molecular homogeneity among polyps from different subjects with hyperplastic polyposis has been noted previously.26
This study also found evidence of molecular heterogeneity among group I subjects. Patient I.7 differed from other group I subjects in having a CIMP-high cancer of the distal colon with MSI-L (instability in MYCL), a CIMP-high SA, and the only example of a HP variant among group I subjects that was also CIMP-high. While falling short of the clinical definition of hyperplastic polyposis,31 the molecular features were unlike those of group I lesions and similar to those of group II lesions. This suggests that hyperplastic polyposis may exist in less penetrant forms that are nevertheless clinically important in terms of cancer risk. Additionally, this case illustrates the fact that lesions with extensive DNA methylation are not restricted to the proximal colon.
Patients with MSI-H cancers frequently have HP but in insufficient numbers to warrant a diagnosis of hyperplastic polyposis. In addition, the HP variant may occur sporadically, outside the condition hyperplastic polyposis.28 Therefore, hyperplastic polyposis, while rare, may represent the most severe manifestation of a predisposition to produce large serrated polyps with extensive DNA methylation and having an increased risk of malignancy.
Dysregulation of methylation seems to act at multiple steps to allow the cell to escape from cell growth control and apoptosis. CIMP is an effective mechanism for gene silencing that does not involve mutations, but none the less produces a stable epigenetic state that is transmitted to daughter cells in a clonal fashion. The concept that hypermethylation occurs on the basis of a pathological lesion has been disputed recently43 but we have shown here that a distinct lesion presenting in hyperplastic polyposis patients exhibits aberrant control of methylation of many genes, including key tumour suppressor genes.
The lack of K-ras mutation in CIMP-high polyps with methylation of hMLH1 was mirrored by the low frequency of K-ras mutations in sporadic MSI-H cancers.11 The latter showed frequent CIMP-high status that included hMLH1 methylation and have been related histogenetically with HP.23,40,44 None of the cancers in this study were MSI-H or showed hMLH1 methylation, even in subjects with hyperplastic polyposis. It is known that cancers in hyperplastic polyposis may be MSI-H, MSI-L, or MSS.23 The common denominator in cancers complicating hyperplastic polyposis may therefore not be the mutator phenotype but a predisposition to DNA methylation that may or may not implicate particular DNA repair genes, such as hMLH1 or MGMT. In this study, all samples of carcinoma in situ or invasive malignancy from patients with hyperplastic polyposis showed at least CIMP-low status and the minimum number of MINTs or genes found to be methylated in this group was three (fig 3). Four of six cancers occurring in group I subjects were MINT-neg and a fifth may have been incorrectly assigned to group I (see above).
Nor is extensive DNA methylation explained merely on the basis of anatomical location. Left sided HP from group II subjects showed more extensive DNA methylation than group I HP. Additionally, it has been pointed out that whereas traditional HP are restricted to the distal colorectum, the HP variant may occur distally as well as proximally.29 It may therefore not be possible to distinguish clinically significant from non-significant serrated polyps on the basis of the number of polyps or anatomical location.
In summary, the findings lend support to the concept of a serrated pathway to CRC in which DNA methylation plays an important role. However, only a subset of serrated polyps is implicated. Among these is a group that is well represented in subjects with hyperplastic polyposis, is characterised by CIMP-high status, methylation of hMLH1, absence of K-ras mutation, and a high frequency of dysplasia. At the morphological level, most of these lesions are equivalent to the recently described “sessile serrated adenomas” or to mixed polyps when dysplastic subclones are present. It is hypothesised that these are the precursors of sporadic MSI-H CRC. Similar lesions but lacking methylation of hMLH1 and having a K-ras mutation may be precursors of CIMP-high CRC that are MSS or MSI-L. ACF and classical HP differ from these lesions in their predilection for the distal colorectum, lower frequency of DNA methylation, and higher frequency of K-ras mutations. The pattern and extent of DNA methylation may determine whether a lesion remains small and clinically insignificant or has the potential for progression. Interpatient differences among subjects with hyperplastic polyposis indicate subtle mechanistic differences that counter the suggestion that DNA methylation occurs exclusively as a stochastic age related process.21 The finding of familial clusters of hyperplastic polyposis26 points to an underlying genetic predisposition to extensive and early onset of DNA methylation.
Acknowledgments
The authors are grateful to the patients who participated in the study and the surgeons who provided specimens. This work was supported by the National Health and Medical Research Council of Australia, the Walter Paulsen Memorial Tumor Bank, and NIH Grant CA77057.