Article Text

Abstract
Objective The antiviral efficacy of nucleos(t)ide analogues whose main limitation is relapse after discontinuation requires long-term therapy. To overcome the risk of relapse and virological breakthrough during long-term therapy, we performed a phase I/II, open, prospective, multicentre trial using a HBV envelope-expressing DNA vaccine.
Design 70 patients treated effectively with nucleos(t)ide analogues for a median of 3 years (HBV DNA <12 IU/mL for at least 12 months) were randomised into two groups: one received five intramuscular injections of vaccine (weeks 0, 8, 16, 40 and 44) and one did not receive the vaccine. Analogues were stopped after an additional 48 weeks of treatment in patients who maintained HBV DNA <12 IU/mL with no clinical progression and monthly HBV DNA for 6 months. The primary endpoint was defined as viral reactivation at week 72 (HBV DNA >120 IU/mL) or impossibility of stopping treatment at week 48.
Results Reactivation occurred in 97% of each group after a median 28 days without liver failure but with an HBV DNA <2000 IU/mL in 33%; 99% of adverse reactions were mild to moderate. Immune responses were evaluated by enzyme-linked immunosorbent spot and proliferation assays: there was no difference in the percentage of patients with interferon-γ secreting cells and a specific T-cell proliferation to HBcAg but not to HBsAg after reactivation in each group.
Conclusions Although it is fairly well tolerated, the HBV DNA vaccine does not decrease the risk of relapse in HBV-treated patients or the rate of virological breakthrough, and does not restore the anti-HBV immune response despite effective viral suppression by analogues.
Trial registration number NCT00536627.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
The aim of HBV treatment is complete viral suppression defined as long-term undetectable serum HBV DNA, leading to a decrease in necroinflammation and fibrosis, and a decrease in hepatitis B-related morbidity and mortality.
-
The antiviral efficacy of nucleos(t)ide analogues is mainly limited by relapse after discontinuation requiring long-term therapy as well as by a potential virological breakthrough during long-term therapy.
-
Although the combination of interferon and nucleo(s)tides does not clearly enhance antiviral potency, therapeutic vaccinations could stimulate a patient's specific immune response and be a promising therapeutic option.
What are the new findings?
-
In this phase I/II, open, prospective, multicentre trial performed in 70 patients with undetectable HBV DNA who had been treated by nucleos(t)ide analogues for a median of 3 years, the use of an HBV envelope-expressing DNA vaccine (five injections):
-
was well tolerated
-
did not decrease the risk of relapse in HBV-treated patients or the rate of virological breakthrough
-
did not restore the anti-HBV immune response despite effective viral suppression by analogues.
-
How might it impact on clinical practice in the foreseeable future?
-
Other immunological strategies should be tested to enhance the long-term efficacy of nucleos(t)ide analogues in patients with chronic hepatitis B, either with other vaccine schedules (number of injections and/or interval between injections, T cell vaccines) or other immunological approaches.
Introduction
Approximately 1 million of the 370 million people with chronic HBV infection die each year from HBV-related liver disease, mainly associated with cirrhosis and hepatocellular carcinoma.1 The goal of therapy is to achieve complete viral suppression (long-term undetectable serum HBV DNA), leading to inactivation of necroinflammation, regression of fibrosis and even the reversal of cirrhosis, significantly decreasing morbidity and mortality.1–3 Nucleos(t)ides analogues (Nucs) result in viral suppression in most adherent patients. First-generation Nucs (lamivudine (LAM) and adefovir dipivoxyl ADV, and telbivudine to a lesser extent) have a low barrier to resistance, explaining the risk of viral resistance, associated with a potential exacerbation and long-term progression to the complications of the highly replicative chronic phase.2 ,4–11 Second-generation Nucs are highly potent and have a high genetic barrier to resistance; they are now the recommended first-line therapy for chronic HBV infection. In addition to concerns about long-term safety, elimination of the covalently closed circular DNA (cccDNA) from HBV-infected hepatocytes is rarely achieved.12 Thus, long-term treatment is necessary except in patients achieving either HBeAg/anti-HBe seroconversion in HBe antigen-positive patients or HBs antigen loss.
Both the adaptive and innate immune responses are known to be involved in viral clearance during acute self-limited HBV infection.13 An alternative approach to antiviral treatment is modulating the defective anti-HBV immune response in patients with chronic infection. Because the combination interferon and nucleo(s)tides did not significantly improve results,14–16 therapeutic vaccination could be promising by stimulating the patient's immune response.17 ,18 Results of therapeutic trials with the HBV protein vaccine were controversial.19 ,20 Recently, DNA vaccination has been shown to increase T cell responses compared with the protein vaccine.21–23 Indeed, the functional profile of the immune response generated by DNA immunisation has been shown to be similar to that obtained in patients with resolved HBV infection and self-limited disease.24 Therefore, since T cell hypo-responsiveness is associated with a high viral load, reducing the viral load before administering the vaccine could improve the immune response.25 ,26 We tested this hypothesis in a randomised study in patients treated with Nucs and undetectable HBV DNA, evaluating the putative additive immune efficacy of a HBV envelope-expressing DNA vaccine.
Materials and methods
Patients
ANRS HB02-VAC-ADN was an open, prospective, phase I/II, randomised, non-blinded, 72-week-trial performed in 19 centres in France. Patients were eligible for inclusion if they were between 18 and 75 years old, with chronic HBV infection defined by a positive hepatitis B surface antigen (HBsAg) for more than 6 months, in whom antiviral treatment had been indicated in an expert centre. Included patients did not have cirrhosis (based on liver biopsy or a non-invasive test for fibrosis performed in the 2 years before the inclusion) or hepatocellular carcinoma (based on an abdominal ultrasound at 6-month intervals). Finally, all included patients had been receiving nucleos(t)ide analogues for at least 3 years with a viremia below 12 IU/mL (corresponding to approximately 60 copies/mL of HBV DNA) for at least 1 year at inclusion, and serum alanine aminotransferase (ALT) below fivefold the upper normal value.
The main exclusion criteria are found in online supplementary 1. Informed consent was obtained from each patient, and the study protocol followed the ethical guidelines of the Declaration of Helsinki and was approved by the human research subjects committee.
Study design
The pCMV-S2.S DNA vaccine produced under good manufacturing practice conditions encodes the small (S) and middle (pre-S2+S) proteins of the HBV envelope.22 The vaccine was stored as 1.0 mL doses at −20°C and thawed at room temperature for 30 min before injection. Randomisation was performed in a centralised procedure. Permuted blocks with a block size of 4 were used, and no stratification was performed. Patients were randomised in a 1:1 ratio to HBV DNA vaccine or no vaccine (control group). Patients randomised in the vaccine group received 1 mg of HBV DNA vaccine at weeks 0, 8, 16, 40 and 44 with intramuscular injections of 0.5 mg in the right deltoid muscle and 0.5 mg in the deltoid muscle at each point. All patients also continued their Nucs treatment up to week 48.
At week 48, antiviral treatment was discontinued if HBV DNA had remained below 12 IU/mL between baseline and week 46, the ALT level was below 10-fold the upper normal value and if liver disease had not progressed. In case of virological breakthrough (defined as the detection of HBV DNA >12 IU/mL before discontinuation of antiviral treatment, confirmed by a second test within 2 weeks) or virological reactivation (defined as a HBV DNA level above 120 IU/mL, confirmed by a second test within 2 weeks) after treatment discontinuation, investigators were advised to switch or to restart antiviral therapy immediately, respectively.
Assessment of efficacy and adverse events
Patients went to follow-up consultations every 4 weeks until week 72, plus a visit at week 46. HBV DNA was determined at each centre at weeks 0, 8, 16, 24, 32, 40, 44, 46, 52, 56, 60, 64, 68 and 72. HBsAg and HBeAg and Ab were measured every 12 weeks. Serum ALT and bilirubin were monitored at each visit and haematological parameters every 12 weeks. Whole blood was collected at weeks 0, 18, 40, 46, 60 and 72 for peripheral blood mononuclear cell (PBMCs) isolation and assessment of T cell immune response to HBcAg and HBsAg.
HBV disease progression was defined as the progression to cirrhosis and/or hepatocellular carcinoma (based on clinical, biological and morphological features, including prothrombin time, albuminemia, alfa-feto protein and abdominal ultrasound examination).
Information on local and general reactions was recorded in the vaccine group 1 h after the vaccination and for 7 days afterwards on diary cards. Safety evaluations included the occurrence and severity of local complications (hard mass, erythema, nodule, pain, pruritus), general reactions (headache, myalgia, arthralgia, pyrexia, asthenia, nausea, vomiting, diarrhoea, rash) and any other adverse events. Adverse events not linked to HBV disease progression or to the vaccination were also recorded.
Primary endpoint
The primary endpoint was virological failure at week 72 (6 months after Nucs discontinuation), defined by either viral reactivation (serum HBV DNA >120 IU/mL) or a virological breakthrough (HBV DNA >12 IU/mL) during Nucs treatment. Both of these events were confirmed by a second test 2 weeks after HBV DNA was first detected. Virological failure also included patients in whom analogue therapy was maintained at week 48 because the liver disease had progressed, defined as progression to cirrhosis and/or its complications: ascites, variceal haemorrhage, encephalopathy and/or hepatocellular carcinoma.
Details of the immunological substudy, immunological assays (HBV antigens and synthetic peptides, proliferation assay, enzyme-linked immunosorbent spot (ELISpot) assay), which has been partially previously described,22 are given in online supplementary 2.
Statistical analysis
With 35 patients per group, it was calculated that the study would have 80% power to detect, a 35% difference in the proportion of virological failures in the two groups using a 5% two-sided test, based on an expected failure rate of 55% in the control group (see online supplementary 3).
Results
Population
In total, 107 patients were initially screened between February 2008 and May 2009; 72 of these were randomised and 70 analysed (34 and 36 patients in the vaccine and the control groups, respectively) (figure 1). Two patients in the vaccine group were excluded, one who withdrew consent before week 0 and another who was incorrectly randomised (mental handicap prohibiting participation).

Enrolment and outcomes in the vaccine randomised trial in HBV patients treated with analogues with undetectable viremia. Antiviral treatment was discontinued at week 48 and serum HBV DNA levels were checked monthly. If viral reactivation (HBV DNA >120 IU/mL, confirmed by a second test within 2 weeks) occurred, patients were asked to restart their antiviral therapy.
Baseline characteristics were similar in the two groups (table 1). Most patients (88.6%) were HBeAg-negative and approximately half had been infected perinatally or in early childhood. Median viremia before Nucs treatment was 6.0 log10 IU/mL in the vaccine group and 6.1 log10 IU/mL in the control group. The duration of Nucs treatment before inclusion in the trial was not significantly different between the vaccine and control groups 3.1 (IQR 2.0–4.6) and 3.0 (IQR 2.3–4.8) years, respectively. Half of the patients (48.6%) were receiving combination therapy at inclusion. HBV DNA was undetectable based on eligibility criteria at the screening and baseline visits. The ALT level was normal in 81% of the patients at inclusion, from onefold to threefold the upper limit of normal in 17% and from threefold to fivefold the upper limit in one patient.
Baseline patient characteristics
One patient received the vaccination subcutaneously and one patient did not receive the last two injections of vaccine due to progression of liver disease (hepatocellular carcinoma).
Serovirological response
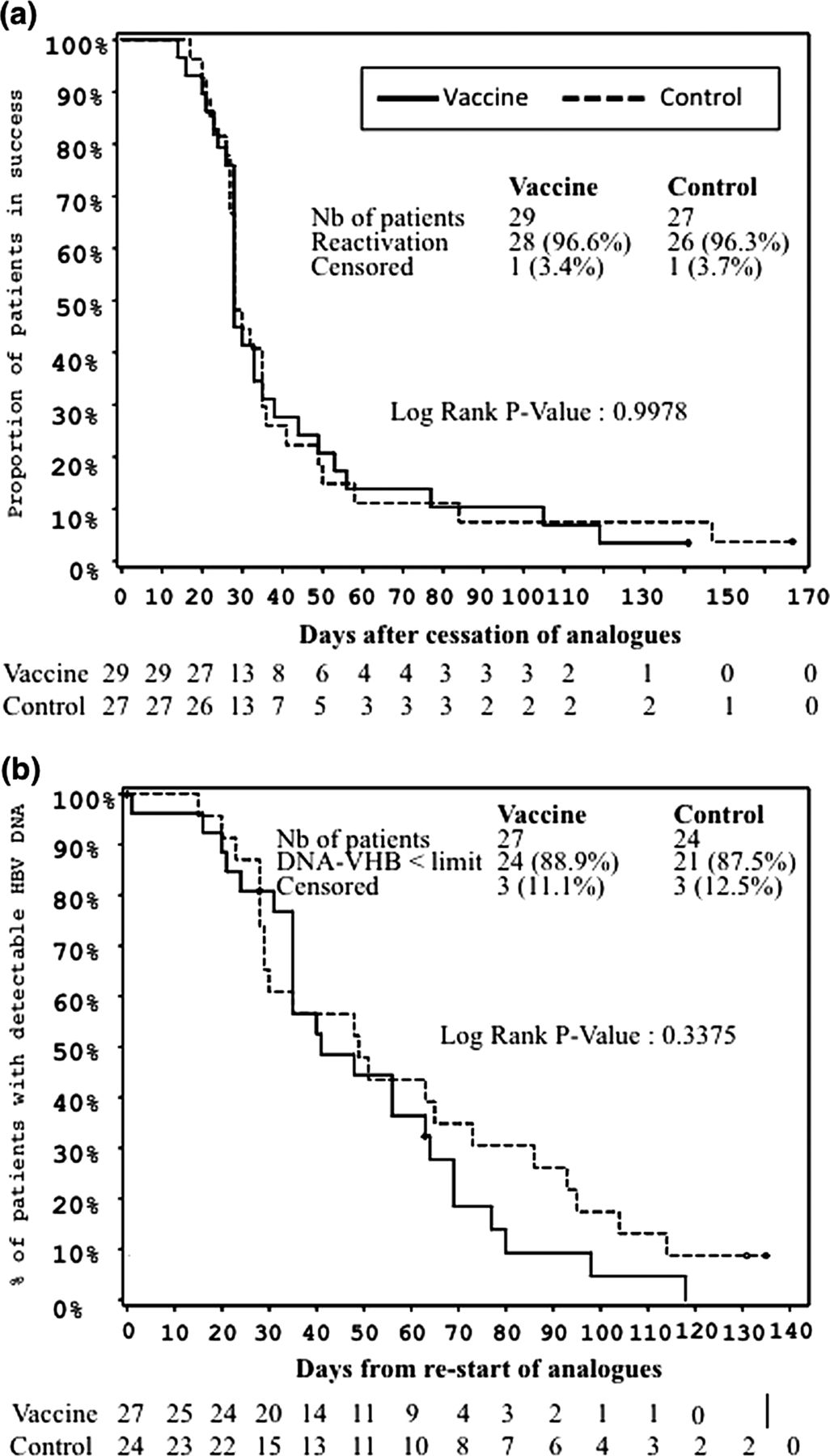
One patient in the vaccine group did not stop Nucs at week 48 due to disease progression (hepatocellular carcinoma). Virological breakthrough occurred in seven patients (four in the vaccine group and three in the control group) after a median 40 weeks and with median HBV DNA levels and ALT level of 73 IU/mL and 28 IU/L (upper normal value <35 IU/L), respectively. Nucs were not stopped, but were modified in these patients according to the protocol, with complete viral suppression in all (see online supplementary 2). Virological failure occurred at week 72 in 33 (97%) and 35 (97%) patients in the vaccine and control groups, respectively (p=1.00) (table 2). Reactivation was observed in 52 patients after treatment discontinuation (27 in the vaccine group, 25 in the control group). After stopping Nucs, the daily increase in viremia was 0.08 log10 IU/mL in both groups (table 2). At reactivation, the median HBV DNA was 2346 IU/mL (997 in the vaccine group and 4150 in the control group) (p=0.30) and 67% of the patients had HBV DNA above 2000 IU/mL. In the 56 patients who stopped therapy, the highest median (min-max) HBV DNA level was 16 805 IU/mL (39–28.000.000) and the viremia was above 2000 IU/mL in 37 (66%). Reactivation also occurred within a median of 28 days (p=1.00; figure 2A) in both groups. At reactivation, median ALT was 29 IU/L (30 in the vaccine group, 29 in the control group; p=0.49). Treatment with Nucs was begun again after a median 21 and 13 days after reactivation in the vaccine and control groups, respectively (p=0.16). The same Nucs were used as before discontinuation at week 48 to treat reactivation in 29 patients.
Virological efficacy of DNA vaccine in vaccine and control groups

Kaplan–Meier: time to HBV viral reactivation after discontinuation of analogue therapy (A); time to HBV-DNA lower than the limit of detection after restart of analogue therapy (B). The number of patients at risk is indicated. Patients from the vaccine group and the control group are represented as continuous and dotted lines, respectively.
Details regarding the Nucs used to treat reactivation and the two patients who had undetectable HBV DNA off therapy are given in online supplementary 4.
At week 72, two patients (one per group) had undetectable HBV DNA off therapy (see online supplementary 4)
The decline in HBsAg titres was not different between week 0 and week 46 in the vaccine group and in the control group, on the one hand, and between the two groups, on the other hand. At week 0, median HBsAg levels were 2224 IU/mL (IQR 1266–4435) in the vaccine group and 2249 IU/mL (IQR 1043–4474) in the control group. Overall 54 (77%) patients had HBsAg levels above 1000 IU/mL. At week 46, median HBsAg levels were 1578 IU/mL (IQR 1008–5170) in the vaccine group and 1912 IU/mL (IQR 977–3693) in the control group. Overall 50 (76%) patients had HBsAg levels above 1000 IU/mL. The median decline in HBsAg levels from week 0 to week 46 was not different between the two treatment groups: −147 in the vaccine group and −220 in the control group (p=0.80). There was no correlation between HBV DNA levels and HBsAg titres.
HBsAg loss occurred in three patients: one patient in the control group at week 60, treated with LAM for 13.4 years (initial HBV DNA not available) with an HBV breakthrough at W44, and without discontinuation of Nucs therapy; one patient in the vaccine group at week 72, treated with ADV for 4.6 years (initial HBV DNA level before Nucs 5000 IU/mL), with HBV reactivation at W52 and re-treated with ADV at W54, with an undetectable HBV DNA at W64; and another patient in the vaccine group at week 72 treated with Nucs for 3.2 years (ADV and LAM at inclusion with a baseline HBV DNA level before Nucs of 6 021 028 IU/mL), HBV reactivation at W56 (997 UI/mL) that was confirmed at W58 (1785 IU/mL) treated with tenofovir disoproxil fumarate (TDF) at W60 (57 UI/mL the same day) and undetectable HBV DNA at W64. The initial HBe status before Nucs use was not available in these three patients, but they were HBeAg(−) and anti-HBeAb(+) at inclusion. Since HBsAg loss occurred either after reactivation or after a virological breakthrough, these three patients were considered to be failures.
Anti-HBs seroconversion occurred in one patient in the control group at week 72, treated with Nucs for 8.6 years (ADV and LAM at inclusion and HBV DNA level before Nucs 271 140 000 IU/mL), with HBV reactivation at W52 (3077 UI/mL) treated with TDF at W52 and undetectable HBV DNA at W56. We did not observe anti-HBs antibodies associated with HBsAg.
Only 8/52 patients were HBeAg(+) at the inclusion: three in the vaccine group and five in the control group. The only significant difference between HBeAg(+) and HBeAg(−) was the total and current treatment duration: 6.0 (3.0–8.2) vs 3.7 (2.1–4.0), respectively (p=0.022).
Immunological response
PBMCs were evaluated in patients for their capacity to secrete IFN-γ in an ex vivo ELISpot assay. At week 0, a positive response against the pre-S2, S and capsid peptides was detectable in 12%, 6% and 18% of patients in the control group and 12%, 9% and 24% of patients in the vaccine group, respectively (figure 3). The percentage of responders varied over time, but the profile remained the same in both groups. In contrast, a small but not significant increase in the median number of IFN-γ-producing T cells was noticed in responders following pre-S2 stimulation from weeks 40 to 72. Between weeks 0 and 40, a median increase of 308 spots/million of PBMC was observed in the responders of the vaccine group compared with 70 spots/million of PBMCs in the responders of the control group (p=0.22; data not shown). A non-significant increase was also found in the vaccine group at week 46 following S stimulation in the vaccine group (a median variation of 235 and 123 spots/million PBMCs in the responders of the vaccine group and control group, respectively; p=0.68). In comparison, the frequency of core-specific T cells remained stable over time (figure 3C).

Frequency of patients with positive responses in ex vivo IFN-γ enzyme-linked immunosorbent spot assay during the trial. Peripheral blood mononuclear cells (PBMCs) from patients were evaluated at different time points for their capacity to secrete IFN-γ after an overnight incubation with pools of overlapping 15-mer peptides covering the pre-S2 (A), S (B) and capsid (C) proteins of HBV. White and black bars represent the percentage of patients with positive responses (% of responders) among the control group and the vaccine group, respectively. The median value (75th centile) of spots/million of PBMC was represented at the right of the figure (* net number of spots after subtraction of background censored at 0 when negative). The number (n) of patients analysed at each time point is indicated beneath the figure.
The cellular immune response was also evaluated by stimulating PBMCs with either HBsAg particles or capsid proteins. Proliferative responses to HBsAg were observed in less than 10% of patients whatever the group or the time of blood collection (figure 4, upper panel). In contrast, 35% and 23% of patients had a proliferative response to HBcAg at week 0 in the vaccine group and the control group, respectively. Following relapse of antiviral treatment at week 48, this proportion increased in both groups (68% vaccine group, 55% control group at week 72) (figure 4, lower panel). There was a significant difference in T cell proliferation in the responders to HBcAg in both groups between week 0 and week 60 or week 72 (Test de Mac-Nemar, p=0.0001). Reincrease in virus particle production could account for this effect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Longitudinal analysis of proliferative responses to HBV antigens. Peripheral blood mononuclear cells from patients were evaluated for their capacity to proliferate following stimulation with either 3 µg/mL of HBsAg particles (A) or 1 µg/mL of capsid proteins (HBcAg) (B) of HBV. White and black bars represent the percentage of patients with positive responses (% of responders) among the control group and the vaccine group, respectively. The median values (and IQR) are represented on the right-side of the figure. The number (n) of patients analysed at each time point is indicated beneath the figure.
No correlation was found between HBsAg level and immunologic data (proliferative response and IFN-γ ELISpot) at week 0.
We studied the link between immunological response at week 60 and the maximum HBV DNA level reached after Nucs discontinuation using the Wilcoxon rank-sum test. The HBsAg proliferative response was too low to study any correlation. For the HBcAg proliferative response, responders at week 60 had significantly higher median maximum HBV DNA levels than the non-responders (58 456 UI/mL vs 1538 IU/mL, p=0.023). For the ELISpot assay at week 60, responders did not have higher median maximum HBV DNA levels than the non-responders (pre-S2: 28 432 IU/mL vs 9606 IU/mL, p=0.65; S: 32 861 vs 9452 IU/mL, p=0.48; capsid: 81 628 vs 6932 IU/mL, p=0.06).
Adverse events
Twenty patients (59%) had at least one local adverse event reported in the vaccine group after injection (78% were mild and 22% moderate), including injection site pain (41%) and injection site induration (21%). Eighteen patients had at least one systemic adverse event (65% mild, 33% moderate and 2% severe): asthenia (50%), myalgia (41%), headache (38%), arthralgia (32%) and nausea (26%).
The proportion of adverse events was similar in both groups through week 72. Fourteen severe adverse effects were reported, but none were vaccine-related: seven hospitalisations (iron-deficiency anaemia, ankle fracture in the vaccine group; sub-ileus, chest pain, urinary tract infection, colonoscopy for screening of polyps, glomus tumour in the control group); one acute hepatitis A; five acute hepatitis (3 and 2 in the vaccine and control group, respectively), which resolved after a re-challenge of new Nucs. Among them, one hepatitis was related to low compliance at week 48 in the vaccine group and four to HBV reactivation after a median 9.5 weeks after discontinuation of Nucs and with a median (min-max) level of ALT of 13-fold (12–32), the upper normal value of ALT. Progression of HBV-related liver disease (hepatocellular carcinoma) occurred in one vaccine patient at week 32. This patient was treated by thermo-ablation without relapse at the end of follow-up.
Discussion
This randomised trial including 70 patients who were receiving long-term treatment of chronic HBV replication with Nucs failed to show that five injections of a DNA vaccine encoding the envelope proteins of HBV had an additive antiviral efficacy in reducing virological breakthrough during Nucs treatment or viral reactivation after the discontinuation of Nucs. In addition, the study of ex vivo peripheral T cell responses did not show any significant vaccine-specific IFN-γ responses during the trial.
These results are disappointing since the synergy between vaccine therapy and analogues could be expected, especially because a specific anti-HBV T cell restoration has been reported with LAM when viral replication decreases.25 Immunomodulatory strategies are needed in the treatment of chronic HBV since the antiviral drugs decrease HBV viremia to undetectable levels by targeting the HBV polymerase, but fail to eradicate infection in most of the patients due to persistent cccDNA in the nuclei of hepatocytes.4–10 Thus, treatment cannot be discontinued because of the risk of reactivation and its associated risks11 ,27 . Long-term or indefinite treatment is necessary but is associated with a risk of resistant strains and potential adverse events, including rare lactic acidosis, nephrotoxicity or loss of bone mineral density.28
In our study, only 2/70 patients had undetectable HBV DNA 24 weeks after discontinuation of Nucs. The higher reactivation rate in the control group than previously reported29 was expected since, when the study was planned, the viral reactivation was defined with a 120 IU/mL threshold for restarting treatment compared with the 2000 IU/mL recommended threshold for treatment initiation in current guidelines.12
By stimulating the patient's functionally impaired immune response, the goal of therapeutic vaccination is to bypass hyporesponsiveness to HBV and reduce the risk of reactivation when Nucs are discontinued, and, to a lesser extent, of virological breakthrough to obtain complete clearance of HBV-infected hepatocytes.30 Several trials of vaccine therapy with HBV protein vaccines have been performed in patients with chronic HBV infection, with discrepant conclusions.19 ,20 ,31 ,32 In these studies, HBV vaccine-specific T cell responses, mostly involving CD4T cells, were induced but without undetectability of HBV DNA in vaccinated compared with control groups. In a large open label, controlled, randomised study, the HBsAg vaccine did not improve the HBe seroconversion rate even in patients treated with LAM who had a vigorous HBsAg-specific lymphoproliferative response, cytokine production and anti-HBs Ab.33 The heterogeneous virological, serological and immunological results probably reflect the heterogeneous patient groups, immunisation procedures and endpoints. Vaccination using immunisation with a DNA-based vaccine elicits antibody responses and T lymphocytes with a Th1 profile. In animal models, including a non-human primate with chronic hepatitis B, intramuscular injection of a plasmid-encoding HBV envelope proteins resulted in a rapid, strong and sustained humoral and cell-mediated immune responses.34–36 In our first phase I trial of therapeutic DNA immunisation using the same DNA vaccine as in this study in patients with LAM resistance, a decrease in viral load and HBeAg seroconversion was observed in 6 and 2/10 patients, respectively. DNA vaccination led to the induction of IFN-γ secreting T cells, demonstrating the ability of this approach to induce or to increase the number of HBV-specific T cells.22 This HBV-specific IFN-γ T cell response was also correlated to an increase in the percentage of a specific NK-cell subset known to produce abundant cytokines, the CD56 bright NK-cell population, but this was not found in the current trial.23
Many factors could play a role in the controversial results with vaccine therapy and in our inability to demonstrate DNA vaccine efficacy. First, our patients were mostly HBeAg(−) with a long history of chronic infection and a disease that was acquired during infancy in half (which is the dominant pattern worldwide37) and in whom functional T cell recovery could only be partial.38 Second, long-term treatment before the study was not associated with a decline in HBsAg titres, which could limit immunological control.39 Third, our DNA vaccine is composed of envelope antigens; the absence of significant changes in ex vivo T cell responses suggests that new formulations, more optimal modes of delivery and/or potent immunogens are needed. Finally, the threshold (imposed by authorities for safety issues) for re-treatment with antivirals was too strict and made it impossible to evaluate sustained impact of vaccine therapy.
These negative results suggest that other immunomodulatory strategies should be evaluated, including other vaccines (T cell vaccine), epitopes or modes of delivery.40 Electroporation-based approaches have been developed in the last 10 years, not only for HBV but also for HIV, HPV and HCV vaccine development. Improvement in the capacity of DNA vaccines to induce T cell responses has been demonstrated in preclinical and clinical settings, but the relevance of this strategy must be validated in patients with chronic infection with HBV.41–43
In conclusion, as revealed by others, despite immune induction and fair tolerance, the virological efficacy of a DNA vaccine was rarely sustained in patients with chronic hepatitis B, with a low rate of HBe seroconversion and HBsAg loss.40 ,41 Even in patients treated with long-term Nucs and without detectable replication, the DNA vaccine did not sufficiently decrease the rate of virological breakthrough during treatment or relapse after analogue discontinuation.
Acknowledgments
The authors thank all the physicians of the ANRS HB02 study group for their active participation in screening, including treating and monitoring the patients: Hôpital Saint Louis, Paris (J.M. Molina); Hôtel Dieu, Lyon (M. Maynard-Huet); Hôpital du Haut Lévêque, Bordeaux-Pessac (P. Couzigou); Hôpital Saint Joseph, Marseille (M. Bourlière); Hôpital de L'Archet, Nice (A. Tran); CHRO La Source, Orléans (X. Causse, S.N. Si Ahmed); Hôpital Avicenne, Bobigny (D. Roulot); Hôpital Jean Verdier, Bondy (N. Ganne-Carrie); CHU de Nancy-Brabois, Nancy (J.P. Bronowicki); CHU Michallon, Grenoble (J.P. Zarski); Hôpital Tenon, Paris (J.D. Grange); CHU Charles Nicolle, Rouen (G. Riachi); CHI Créteil, Créteil (I. Rosa); CHG Victor Jousselin, Dreux (A. Landau); CHU Côte de Nacre, Caen (T. Dao); Hôpital Cochin, Paris (P. Sogni, V. Mallet, A. Vallet-Pichard). The authors also thank O. Launay, M.L. Chaix, F. Degos, ANRS hepatitis team (K. Kaabeche, V. Petrov-Sanchez), E. Mottez and C. Guérin, for participating in the scientific committee, and Drs B. Balkau, J.C. Duclos-Vallée, A.M. Roques-Afonso, D. Samuel and J.P. Viard for participating in the Data and Safety Monitoring Board. Thanks also to the following for participating in the coordinating centre: INSERM SC10, Villejuif, C. Capitant, J. Garrabey, S. Circosta, S. Laffont, C. Lascoux, M. Saouzanet, C. Durier, A. Arulananthan (data management).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
SK and CC contributed equally.
-
Contributors All authors made contributions to the article.
-
Funding The trial was sponsored and funded by The National Agency for Research on Aids and Viral Hepatitis.
-
Competing interests HF: oral communications for Bristol-Myers Squibb, Gilead, Roche, Schering-Plough/Merck, Janssen and participation for medical congress as auditor. SP has received consulting and lecturing fees from Bristol-Myers Squibb, Boehringer Ingelheim, Tibotec, Vertex, Gilead, Roche, Schering-Plough/Merck, Novartis, Abbott, Sanofi and Glaxo Smith Kline, and grants from Bristol-Myers Squibb, Gilead, Roche and Merck/Schering Plough.
-
Patient consent Obtained.
-
Ethics approval Informed consent was obtained from each patient and the study protocol, conformed to the ethical guidelines of the Declaration of Helsinki, was approved by the human research subjects committee (NCT00536627).
-
Provenance and peer review Not commissioned; externally peer reviewed.