Article Text

Abstract
Objective Helicobacter pylori strains that express the oncoprotein CagA augment risk for gastric cancer. However, the precise mechanisms through which cag+ strains heighten cancer risk have not been fully delineated and model systems that recapitulate the gastric niche are critical for understanding pathogenesis. Gastroids are three-dimensional organ-like structures that provide unique opportunities to study host-H. pylori interactions in a preclinical model. We used gastroids to inform and direct in vitro studies to define mechanisms through which H. pylori modulates expression of the cancer-associated tight junction protein claudin-7.
Design Gastroids were infected by luminal microinjection, and MKN28 gastric epithelial cells were cocultured with H. pylori wild-type cag+ strains or isogenic mutants. β-catenin, claudin-7 and snail localisation was determined by immunocytochemistry. Proliferation was assessed using 5-ethynyl-2′-deoxyuridine, and levels of claudin-7 and snail were determined by western blot and flow cytometry.
Results Gastroids developed into a self-organising differentiation axis and H. pylori induced mislocalisation of claudin-7 and increased proliferation in a CagA- and β-catenin-dependent manner. In MKN28 cells, H pylori-induced suppression of claudin-7 was regulated by β-catenin and snail. Similarly, snail expression was increased and claudin-7 levels were decreased among H. pylori-infected individuals.
Conclusions H. pylori increase proliferation in a strain-specific manner in a novel gastroid system. H. pylori also alter expression and localisation of claudin-7 in gastroids and human epithelial cells, which is mediated by β-catenin and snail activation. These data provide new insights into molecular interactions with carcinogenic potential that occur between H. pylori and epithelial cells within the gastric niche.
- HELICOBACTER PYLORI
- GASTRIC CANCER
- CELL PROLIFERATION
- TIGHT JUNCTION
- GASTRIC INFLAMMATION
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Helicobacter pylori is the strongest known risk factor for gastric cancer, yet most infected persons do not develop this malignancy.
-
Disease risk is dependent upon strain-specific virulence factors, host responses to such constituents and environmental factors.
-
Mechanistic studies of H. pylori pathogenesis are limited by current microbial-epithelial systems.
What are the new findings?
-
Murine gastroids can develop into a self-organising differentiation axis and be successfully infected with pathogenic H. pylori strains.
-
H. pylori increase proliferation in gastroids, which is dependent upon the bacterial oncoprotein CagA and β-catenin signalling.
-
H. pylori induce mislocalisation and suppression of the tight junction protein claudin-7, which is regulated by β-catenin and snail.
-
Snail expression is increased and claudin-7 levels are decreased among H. pylori-infected individuals.
How might it impact on clinical practice in the foreseeable future?
-
The use of biologically relevant model systems such as gastroids may delineate novel pathways through which H. pylori lowers the threshold for disease, leading to the identification of new therapeutic targets for antimicrobial therapies.
-
H. pylori infection is inversely related to other diseases, such as oesophageal adenocarcinoma and atopic disorders; therefore, identification of factors that mediate H. pylori-induced gastric cancer may allow physicians to more sharply focus test and treat strategies on high-risk populations.
-
Defining mechanisms through which H. pylori induces gastric cancer may provide important insights into other malignancies that arise from inflammatory foci.
Introduction
Helicobacter pylori colonises the gastric mucosa of over half the world's population and persistent infection significantly increases the risk of developing niche-specific diseases, such as gastric adenocarcinoma.1 Bacterial virulence constituents exert important influences in determining the outcome of infection and H. pylori strains that possess a functional cag pathogenicity island (PAI) incur a higher risk for cancer than cag− strains.2 The cag PAI is comprised of genes that encode components of a type IV secretion system, which translocate bacterial products such as CagA, into host cells following microbial contact.3–6 Intracellular CagA is rapidly phosphorylated by Src and Abl kinases and phosphorylated CagA activates a host phosphatase (SHP-2) leading to changes in cell motility and proliferation.4–7 Unphosphorylated CagA activates β-catenin and can disrupt cell–cell junctions and contribute to loss of epithelial barrier function.8–10
β-catenin is complexed at the cellular membrane in the adherens junction and/or sequestered in the cytosol within a multiprotein inhibitory complex containing adenomatous polyposis coli (APC), glycogen synthase kinase (GSK)-3β, and axin.11 This complex constitutively targets β-catenin for proteosomal degradation via GSK-3β-mediated phosphorylation. H. pylori activates β-catenin through inactivation of GSK-3β, or via an interaction between CagA and membrane-associated β-catenin, which promotes mitogenic signalling and proliferation.9 ,12 ,13
Another apical-junctional protein that may influence H. pylori-induced proliferation in conjunction with β-catenin is claudin-7. Claudin-7 is a member of a family of 24 transmembrane proteins, which are important constituents of tight junctions and which are the main determinants of tight junction barrier function.14 Disruption of tight junction complexes is associated with a variety of human diseases, including cancers of the GI tract.14 H. pylori adhere to gastric epithelial cells in close proximity to tight junctions and can alter localisation of the component proteins that constitute these complexes.15 ,16 Altered expression of claudin-7 has been implicated in several types of human cancers,14 and in a mouse model of intestinal claudin-7 deficiency, loss of claudin-7 leads to increased proliferation.17
An upstream mediator of several claudins is snail, a member of the zinc-finger family of transcription factors. Snail functions as a transcriptional repressor and can repress E-cadherin through binding to E-boxes within the E-cadherin promoter; of note, increased snail expression is also associated with malignancy.18 Snail suppresses the expression of claudins19 and, specifically, claudin-7 is negatively regulated by snail in vitro.18 Concordant with these data, H. pylori can upregulate snail, leading to epithelial:mesenchymal transition.20
Investigations to identify mechanisms by which H. pylori induces aberrant gastric epithelial responses have been primarily driven by in vitro cancer cell models, short-term ex vivo primary cell models or in vivo rodent models of infection. However, detailed studies of interactions between H. pylori and intact gastric epithelium have been limited by issues of in vivo accessibility or dedifferentiation in cell culture.21 ,22 Therefore, the aim of this study was to use a novel replenishable ex vivo three-dimensional (3D) system to identify constituents that mediate host–H. pylori interactions with carcinogenic potential and to subsequently inform in vitro mechanistic studies that could ultimately be extended into human gastric epithelium. Our data show that H. pylori induce proliferation in gastroids and that this is dependent upon the H. pylori virulence factor CagA and β-catenin. Additionally, H. pylori alter the expression of claudin-7 in human gastric tissue and in gastric epithelial cells via signalling pathways that involve β-catenin and snail.
Materials and methods
Details for cell culture, western blot analysis, transfections and luciferase assays, transient transfection of siRNA and immunofluorescence are contained in the online supplementary data.
Animals and gastroid culture
All procedures were approved by the Institutional Animal Care Committee of Vanderbilt University. C57BL/6 mice were euthanased at 8–12 weeks of age. Mouse stomachs were ligated at the oesophago-gastric and gastric-duodenal junctions, rapidly removed and washed in phosphate buffered saline (PBS). Approximately half the forestomach was removed. The pyloric sphincter was then directed through the newly created fundic opening, and the stomach inverted and sealed by ligation of the remaining forestomach. Inverted stomachs were washed in ice-cold Dulbeccos PBS (DPBS), filled by injection with DPBS and incubated at 4°C in EDTA (5 mM) for 2.5 h. EDTA was then replaced with 5 mL of osmotic shaking buffer (54.9 mM D-Sorbitol and 43.5 mM sucrose) and stomachs were shaken to remove glands. Released glands were centrifuged at 250g for 10 min and glands were plated in Matrigel (BD Biosciences) containing N-acetylcysteine (1 μM), gastrin (10 nM), epidermal growth factor 10 (50 ng/mL), R-spondin 1 (500 ng/mL), Noggin (100 ng/mL), fibroblast growth factor 10 (100 ng/mL), rWnt3A (100 ng/mL) and Y27632 (10 μM), based on recently published protocols developed for gastroid cultures.21 Advanced Dulbecco's Modification of Eagle's Medium (DMEM)/F12 medium supplemented with B27, N2, penicillin/streptomycin, N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (10 mM) and Glutamax (2 mM) was overlaid on the Matrigel. For coculture experiments with H. pylori, advanced DMEM/F12 medium without penicillin/streptomycin was used.
H. pylori strains and culture conditions
The H. pylori cag strains 7.13 or 60190 were grown in Brucella broth with 10% fetal bovine serum for 16 h, harvested by centrifugation and cocultured with MKN28 cells or microinjected into the lumen of gastric gastroids at a multiplicity of infection of 100:1.8 ,23 An isogenic cagA null mutant constructed in strain 7.13 was selected with kanamycin (25 μg/mL) as previously described.8
Flow cytometry
Gastric epithelial cells were isolated from frozen human gastric biopsies using a dissociation and dispersion technique, as previously described.24 ,25 Gastric biopsies were obtained from H. pylori-infected or uninfected patients residing in Colombia,26 which was approved by the ethics committees of the participating hospital and the Universidad del Valle in Cali, Colombia, and the institutional review board of Vanderbilt University. Gastric tissue was treated with 10 µM dithiothreitol at room temperature for 30 min and 1.0 mM EDTA for 30 min at 4°C. Dispersed cells were filtered through a 70 µm filter (BD Falcon) to isolate single cells. Cells were fixed and permeabilised with 0.1% paraformaldehyde and ice-cold methanol. Cells were then incubated with a mouse monoclonal anti-pan-cytokeratin antibody conjugated with allophycocyanin (1:100, BD Biosciences) and a rabbit polyclonal anti-claudin-7 antibody (1:100, Life Technologies) at room temperature for 20 min. Cells were washed and stained with hamster anti-rabbit secondary antibody conjugated with fluorescein isothiocyanate (1:400, BD Biosciences). Cells were acquired using a LSR II Flow Cytometer (BD Biosciences), and pan-cytokeratin-positive cells were analysed for claudin-7 expression using FlowJo (Tree Star).
Analysis of cell proliferation
Cell proliferation was determined 96 h after H. pylori microinjection using 5-ethynyl-2′-deoxyuridine (EdU) assays and analysed by the Click-iT EdU imaging kit (Molecular Probes) according to the manufacturer's instructions. EdU was added to organoids immediately following microinjection of H. pylori.
Immunohistochemistry on human gastric mucosa
Immunohistochemistry (IHC) was performed on paraffin-embedded biopsy samples from patients with or without H. pylori infection. Tissue samples were deparaffinised and stained with a polyclonal anti-snail antibody. A single pathologist (MBP) scored cytoplasmic and nuclear snail IHC staining by assessing the percentage of snail+ epithelial cells semiquantitatively, as previously described.25
Statistical analysis
Results are expressed as means±SEM. Comparisons were made using the Mann–Whitney student t test or analysis of variance, and were considered significant at p≤0.05.
Results
H. pylori alters β-catenin and enhances proliferation in gastroids
H. pylori activates β-catenin signalling in transformed gastric epithelial cells8–10; however, the specific effectors and cellular phenotypes regulated by β-catenin that contribute to carcinogenesis have not been fully defined. Gastroids are 3D model systems that more closely recapitulate events occurring within gastric epithelium than traditional clonal cell culture systems,27 thereby providing new opportunities for the molecular dissection of epithelial responses to microbial pathogens. As a prelude to defining pathways altered by carcinogenic H. pylori strains ex vivo, we generated gastroids from mice and examined lineage allocation. These results indicated that gastroids self-organise into a sphere of cells surrounding a central lumen and differentiate into mucus cells, parietal cells, G-cells, enterochromaffin-like (ECL) cells (figure 1A–G) and D-cells (not shown). We subsequently determined that gastroids could be successfully infected with H. pylori via microinjection into the organoid lumen, with bacterial viability lasting for up to 7 days (figure 1H). To determine if gastroids accurately reflect in vivo responses to H. pylori, we examined occludin localisation. As determined by immunofluorescence, H. pylori infection of gastroids resulted in disruption of occludin localisation; importantly, these findings replicate our previously published in vivo findings in H. pylori-infected insulin-gastrin (INS-GAS) mice that resulted in mislocalisation of occludin at the tight junction.16
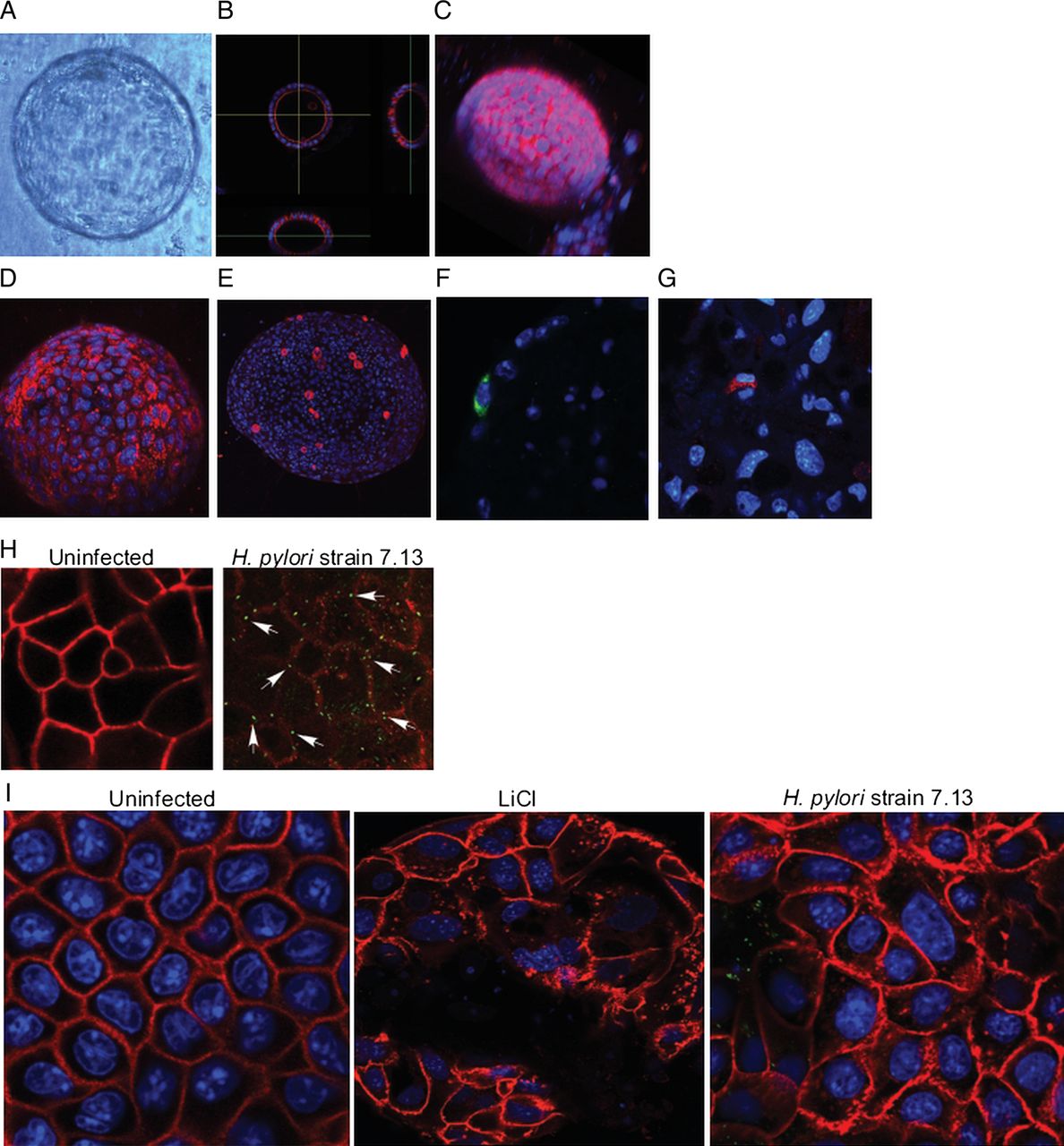
Gastroids differentiate and can be successfully infected with Helicobacter pylori. (A) Bright field image of a single gastroid. (B) Cross-sectional image of a single gastroid stained for F-actin (red); nuclei are labelled with Hoechst (blue). (C) Three dimensional reconstruction of a single gastroid stained for F-actin (red) and nuclei (blue). (D-G) Representative staining of gastroids for (D) gastric mucin (red), labelling mucus cells; (E) H+K+ATPase (red), labelling parietal cells; (F) gastrin (green), labelling G-cells and (G) chromogranin (red), labelling enterochromaffin-like cells. (D–G), nuclei are labelled with Hoechst (blue). (H) Uninfected (left panel) and infected (right panel) gastroids stained for occludin (red) and H. pylori (arrows, green). (I) Gastroids were uninfected, treated with LiCl or infected with H. pylori strain 7.13 (green) and stained for β-catenin (red).
Having established that gastroids could be successfully infected and reflect in vivo findings, we next examined the ability of H. pylori to affect β-catenin, as this molecule orchestrates aberrant epithelial responses with carcinogenic potential. As determined by immunofluorescence, β-catenin sharply localised to cellular margins in uninfected organoids (figure 1I). Lithium chloride (LiCl) activates β-catenin via phosphorylation and inhibition of GSK-3β, and treatment of gastroids with LiCl significantly increased the cytoplasmic pool of β-catenin (figure 1I, see online supplementary figure S1A). Importantly, infection with H. pylori strain 7.13 similarly altered the distribution of β-catenin, leading to an accumulation in the cytosol (figure 1I). To ensure that these findings were not secondary to apoptosis, we treated gastroids with LiCl or infected them with H. pylori strain 7.13 via luminal microinjection and quantified apoptosis using an antibody directed against cleaved caspase 3 (see online supplementary figure S1B, C). Apoptotic cells were only detected in the lumen of gastroids, and levels of apoptosis were not significantly different between uninfected, LiCl-treated or H. pylori-infected gastroids (see online supplementary figure S1B, C).
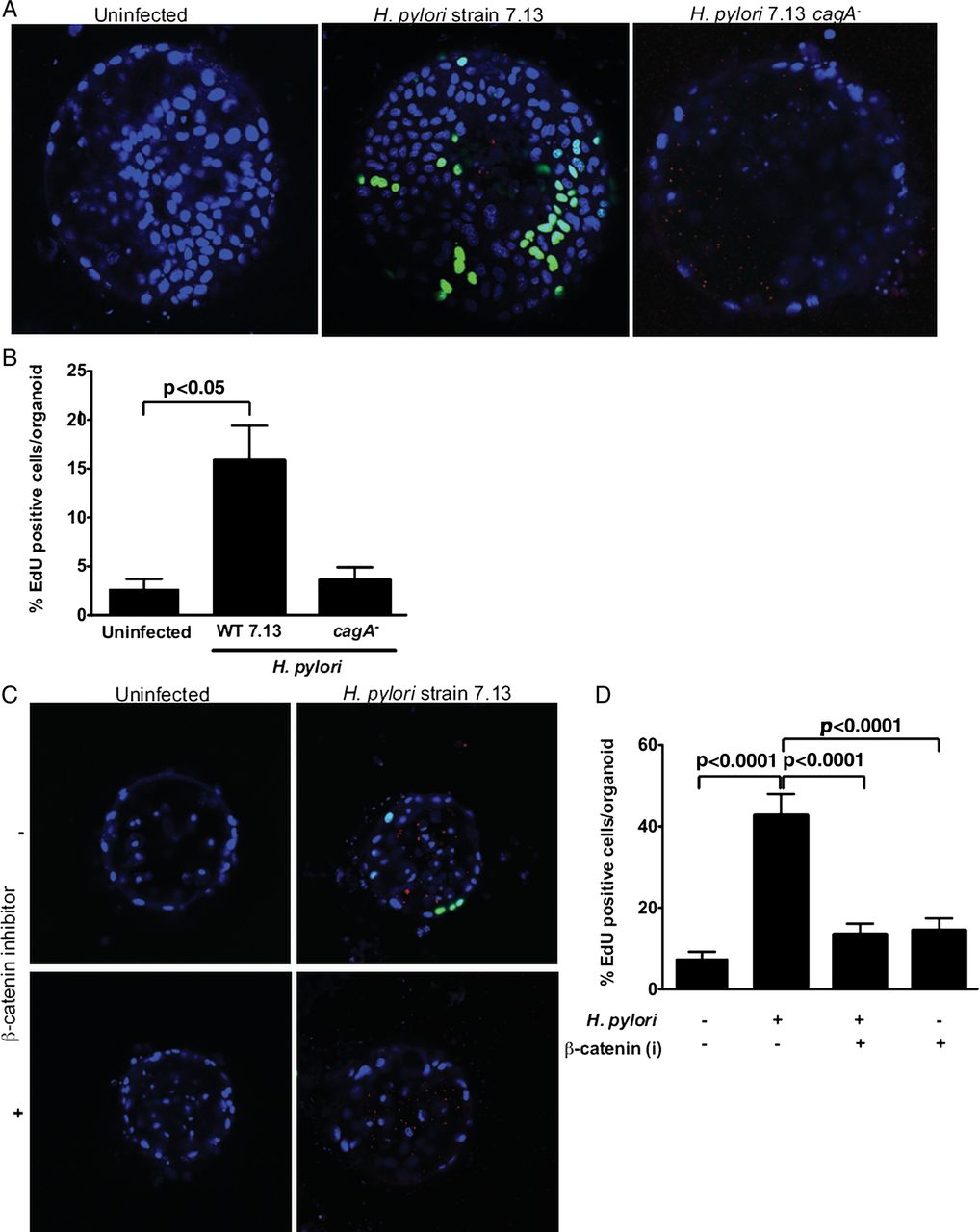
H. pylori strains that express and translocate CagA augment gastric cancer risk and CagA can regulate β-catenin8; therefore, we examined CagA-dependent phenotypes in gastroids that may be linked to carcinogenesis. Gastroids were infected with H. pylori strain 7.13 or an isogenic 7.13 cagA− null mutant via luminal microinjection, and proliferation was quantified using EdU 96 h after H. pylori infection (figure 2A, B). Gastroids infected with wild-type H. pylori proliferated at a significantly higher rate than uninfected organoids. By contrast, proliferation rates of gastroids infected with an isogenic H. pylori cagA− mutant were no different compared with uninfected gastroids (figure 2A, B).

Helicobacter pylori induces proliferation in gastroids via CagA and β-catenin. (A and B) Gastroids were infected with wild-type (WT) H. pylori strain 7.13 or an isogenic 7.13 cagA− null mutant (red) via luminal microinjection and proliferation was quantified using 5-ethynyl-2′-deoxyuridine (EdU) (green). (A) Representative staining in uninfected (left panel), WT H. pylori-infected (middle panel) or H. pylori cagA− mutant-infected (right panel) gastroids. (B) EdU quantification demonstrating increased proliferation in WT H. pylori infected gastroids. (C and D) Gastroids were pretreated with cardionogen 1, a β-catenin inhibitor (i), and then infected with H. pylori and proliferation was quantified by EdU. (C) Representative staining in uninfected or WT H. pylori-infected gastroids, with or without pretreatment with cardionogen 1. (D) EdU quantification demonstrating that increased proliferation in H. pylori WT-infected gastroids is abolished by pretreatment with cardionogen 1. Data are expressed as means±SEM, n=3 independent replicates.
To directly determine whether enhanced proliferation was dependent on H. pylori-induced β-catenin activation, gastroids were pretreated with cardionogen 1, an inhibitor of β-catenin, and then infected with H. pylori. As expected, H. pylori significantly increased proliferation in mock-treated compared with uninfected gastroids; however, pretreatment with cardionogen 1 significantly (p<0.0001) decreased proliferation in infected gastroids, indicating that H. pylori-induced proliferation in this model system is regulated by both CagA and β-catenin (figure 2C, D).
A cancer-associated tight junction protein is aberrantly altered during H. pylori infection of gastroids and gastric epithelial cells
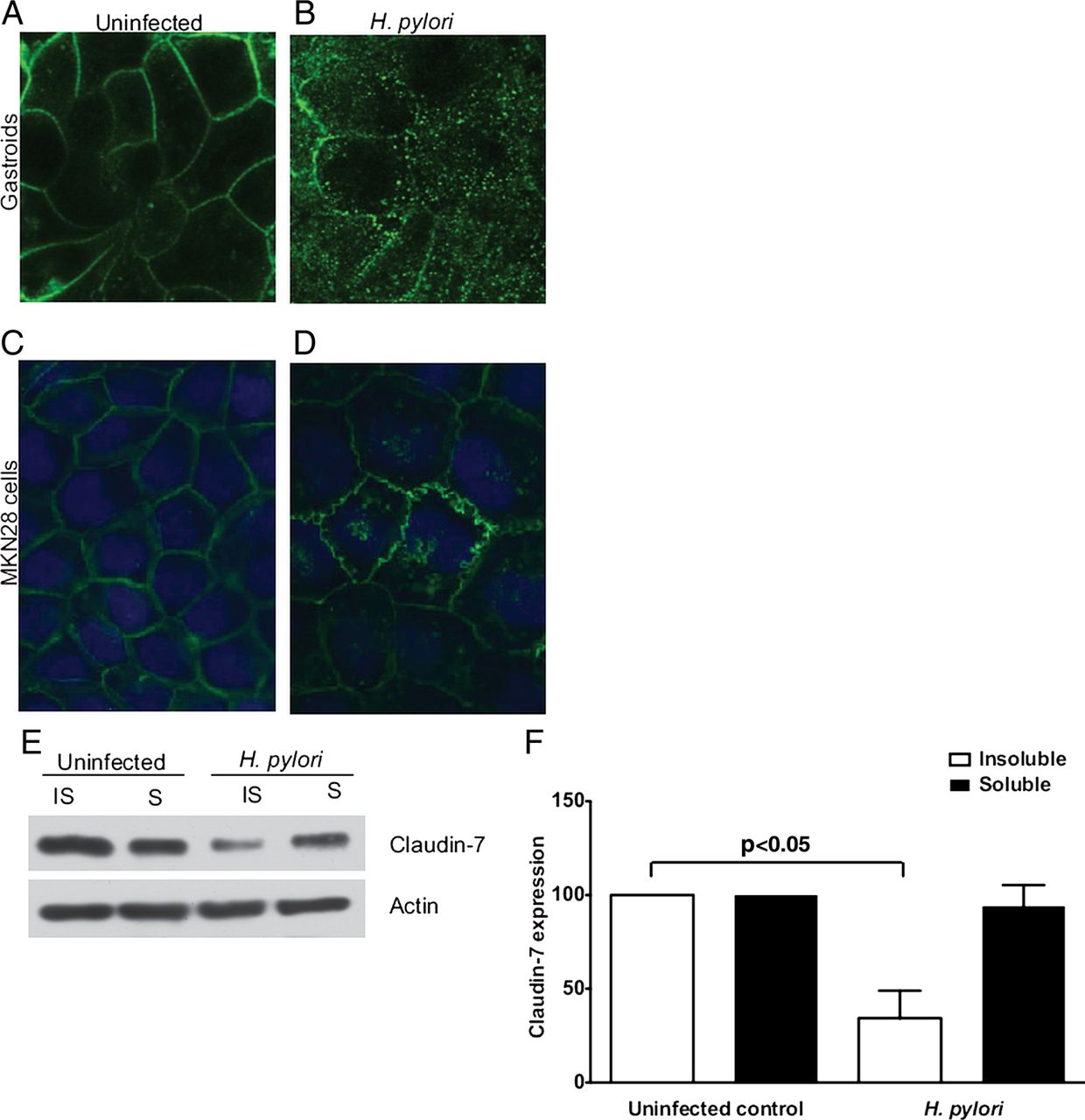
Increased proliferation develops within the intestinal epithelium of claudin-7-deficient mice, and levels of claudin-7 are reduced in several types of cancer14 ,17; therefore, we next examined whether H. pylori altered claudin-7. In uninfected gastroids, claudin-7 was sharply localised to the cellular margins (figure 3A). By contrast, H. pylori infection induced an aberrant redistribution of claudin-7, with focal accumulation in the cytosol (figure 3B). This was not due to apoptosis as cleaved caspase 3 was only present in cells extruded into the gastroid lumen and levels of cleaved caspase 3 were not significantly different between uninfected gastroids or H. pylori-infected gastroids (see online supplementary figure S1B, C).

Helicobacter pylori alters the topography of claudin-7 in gastroids and MKN28 gastric epithelial cells. Immunocytochemical localisation of claudin-7 in uninfected (A) gastroids and (C) MKN28 cells and H. pylori-infected (B) gastroids and (D) MKN28 cells, demonstrating accumulation of claudin-7 in cytosolic vesicles and discontinuous claudin-7 staining at the cell membrane in infected samples. (E and F) MKN28 cells were cocultured for 48 h with H. pylori strain 60190 and claudin-7 levels were determined by western blotting. (E) Representative western blot for claudin-7 or actin in detergent-insoluble and detergent-soluble MKN28 fractions in the presence or absence of H. pylori. (F) Densitometric analysis demonstrating loss of claudin-7 from the insoluble fraction of H. pylori-infected cells. Data are expressed as means±SEM, n=3 independent replicates.
We next sought to confirm and extend results from gastroids into a system that can be easily manipulated, MKN28 human gastric epithelial cells. MKN28 cells and the H. pylori cag strain 60190 were used for these studies because we have previously shown that MKN28 cells form functional apical-junctional complexes characteristic of epithelial cells and that strain 60190 dysregulates tight junctions in this model.16 MKN28 cells were cocultured with H. pylori for 48 h and claudin-7 was examined by immunofluorescence. Similar to findings in gastroids, claudin-7 was localised to the cell membrane in uninfected cells (figure 3C). The topography of claudin-7, however, was disrupted by H. pylori (figure 3D), findings that mirrored our results in gastroids. To confirm these results, western immunoblotting was performed using uninfected and H. pylori-infected MKN28 sub-cellular fractions. Immunoblotting for claudin-7 using soluble (cytosolic) and insoluble (membrane-associated) epithelial cell fractions confirmed redistribution of claudin-7, with a significant (p<0.05) reduction of claudin-7 in the insoluble fraction of infected cells (figure 3E, F).
H. pylori-mediated suppression of claudin-7 is dependent upon β-catenin
Having established the effects of H. pylori on both β-catenin and claudin-7, we next determined whether a direct linkage was present between these host effectors within H. pylori-infected cells. MKN28 cells were transfected with a β-catenin reporter containing three tandem lymphoid enhancer factor/T-cell factor (LEF/TCF)-binding motifs upstream of the luciferase gene (Topflash) or a control construct containing mutant LEF/TCF sites (Fopflash), and then treated with LiCl. As expected, treatment of transfected cells with LiCl increased luciferase activity (figure 4A), reflecting β–catenin activation. We next determined whether β-catenin activation regulated expression of claudin-7. MKN28 cells were incubated with LiCl for 24 h, which led to significantly decreased expression levels of claudin-7 (figure 4B, C). To determine whether H. pylori activation of β-catenin-reduced levels of claudin-7, siRNA targeting β-catenin was used. The decrease in expression of claudin-7 following infection with H. pylori was abolished in infected MKN28 cells treated with β-catenin-specific siRNA (figure 4D, E). These results implicate β-catenin as a key mediator of claudin-7 suppression within the context of H. pylori infection.

Helicobacter pylori-mediated suppression of claudin-7 is dependent on β-catenin. (A) MKN28 cells were transfected with a β-catenin reporter containing three tandem lymphoid enhancer factor/T-cell factor (LEF/TCF)-binding motifs upstream of the luciferase gene (Topflash) or a control construct containing mutant LEF/TCF sites (Fopflash), and then treated with 10 mM LiCl. Luciferase activity, reflecting β–catenin activation, was quantified. (B and C) MKN28 cells were incubated with LiCl for 24 h, and expression levels of claudin-7 or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were quantified by western blotting. (B) Representative western blot for claudin-7 and GAPDH in MKN28 cells in the presence or absence of LiCl. (C) Densitometric analysis demonstrating decreased expression of claudin-7 in LiCl-treated cells. (D and E) MKN28 cells were transfected with control or β-catenin-specific siRNA and levels of claudin-7 or GAPDH were quantified by western blotting. (D) Representative western blot for claudin-7 and GAPDH in MKN28 cells in the presence of non-targeting (NT) or β-catenin-specific siRNA, with or without H. pylori strain 60910. (E) Densitometric analysis demonstrating that decreased expression of claudin-7 in H. pylori-infected cells is abolished in the presence of β-catenin-specific siRNA. Data are expressed as means±SEM, n=3 independent replicates.
H. pylori-induced suppression of claudin-7 is mediated by snail
Snail is a transcription factor that negatively regulates claudin-7 expression in human cancers18; therefore, we next determined if H. pylori-induced downregulation of claudin-7 was mediated by snail in gastric epithelial cells. In MKN28 cells cocultured with H. pylori, snail mRNA expression was significantly increased compared with uninfected cells (figure 5A), which was accompanied by increased protein levels in whole-cell lysates (figure 5B, C). These results were confirmed using immunofluorescence, which demonstrated an accumulation of snail in the nuclei of H. pylori-infected cells (figure 5D). To definitively link H. pylori-induced activation of snail and reduced expression of claudin-7, siRNA was used. Decreased expression of claudin-7 following infection with H. pylori was abolished by snail knockdown, indicating that activation of snail by H. pylori decreases claudin-7 expression (figure 5E,F).

Helicobacter pylori-induced suppression of claudin-7 is mediated by snail. MKN28 cells were cocultured with H. pylori strain 60 190 and levels of snail mRNA (A), and snail total protein (B and C) were quantified. (A) Real-time RT-PCR was performed on isolated RNA and quantified. (B) Representative western blot for snail and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in MKN28 cells in the presence or absence of H. pylori. (C) Densitometric analysis demonstrating increased expression of snail in H. pylori-infected cells. (D) Immunofluorescence for snail (green) in uninfected MKN28 cells or MKN28 cells infected with H. pylori strain 7.13. Nuclei are labelled with Hoechst (blue). (E and F) Control non-targeting (NT) siRNA or siRNA targeting snail was transfected in MKN28 cells followed by infection with or without H. pylori. Levels of claudin-7 expression were then determined by western blotting. (E) Representative western blot for claudin-7 and GAPDH in MKN28 cells in the presence or absence of non-targeting or snail-specific siRNA. (F) Densitometric analysis demonstrating that decreased expression of claudin-7 in H. pylori-infected cells is abolished in the presence of snail-specific siRNA, n=3 independent replicates.
H. pylori infection leads to decreased claudin-7 and increased snail expression in human gastric epithelial cells in vivo and in gastroids
To extend our results into the natural niche of H. pylori, we examined claudin-7 expression in human gastric biopsy specimens by flow cytometry (figure 6A, B). Epithelial cells were selected using a pan-cytokeratin antibody and claudin-7 expression was quantified in the epithelial cell-enriched population. Concordant with our gastroid and cell culture data, expression of claudin-7 was decreased in gastric epithelial cells harvested from infected persons, when compared with uninfected gastric biopsies (figure 6A, B). Having shown that H. pylori also increase snail expression in vitro, we quantified snail expression in human gastric biopsy specimens. Similar to our in vitro results, snail expression was increased in gastric epithelium harvested from persons infected with H. pylori compared with uninfected persons (figure 6C, D, E). Finally, we returned to our novel gastroid model and examined snail expression in uninfected and H. pylori-infected gastroids (figure 6F). Concordant with our in vivo findings, snail expression was markedly increased in the cytoplasm of epithelial cells in H. pylori-infected compared with uninfected gastroids (figure 6F). These results suggest that alterations in claudin-7 and snail may contribute to the ability of H. pylori to induce injury within the human gastric niche.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Helicobacter pylori infection leads to decreased claudin-7 and increased snail expression in human gastric epithelial cells in vivo and in gastroids. (A and B) Claudin-7 expression in human gastric epithelial cells isolated from gastric biopsies from uninfected and H. pylori-infected subjects was assessed by flow cytometry analysis. (A) Representative histogram for claudin-7 in uninfected and H. pylori-infected cells. (B) Levels of claudin-7 protein expressed as MFU determined by flow cytometry, were compared between uninfected and H. pylori-infected samples. (C–E) Snail expression in human gastric tissue was assessed by immunostaining in uninfected (C), and H. pylori-infected subjects (D) at 200× magnification. (E) A single pathologist, blinded to treatment groups, assessed and scored snail immunostaining. Snail IHC score was determined by assessing the percentage of snail epithelial cells multiplied by the intensity of epithelial snail staining (1–3) in both the cytoplasm and nucleus of gastric epithelial cells. Data are expressed as means+SEM. (F) Representative staining for snail (green) in uninfected or H. pylori-infected gastroids; nuclei are labelled with Hoechst (blue). APC, adenomatous polyposis coli; MFU, mean fluorescence units; IF, immunofluorescence; IHC, immunohistochemistry.
Discussion
Gastric adenocarcinoma is the third leading cause of cancer-related death in the world, and approximately 660 000 new cases of gastric cancer/year are attributable to H. pylori, making this pathogen the most common infectious agent linked to malignancy.28 However, only a percentage of colonised persons develop neoplasia, and enhanced risk is related to H. pylori strain differences, host responses governed by genetic diversity and/or specific interactions between host, microbial and environmental determinants.29 Universal test and treat strategies for H. pylori are not feasible due to the high prevalence of infection as well as the expense and side effects of antibiotic therapy.2 ,30 Additionally, evidence indicates that carriage of certain strains is inversely related to oesophageal adenocarcinoma and atopic diseases2 ,30 ,31; thus, investigations directed at understanding mechanisms that regulate malignant progression are required to institute programmes of targeted therapy.
β-catenin is a ubiquitously expressed protein that performs distinct roles within host cells. Our laboratory and other groups have previously demonstrated that CagA activates phosphoinositide-3 (PI3) kinase,12 which inhibits the kinase activity of GSK-3ß,13 leading to inhibition of β-catenin degradation; intracellular CagA also disrupts E-cadherin-β-catenin complexes.9 These events lead to β-catenin nuclear accumulation, resulting in transcription of genes implicated in carcinogenesis. Increased β-catenin expression as well as mutations within APC are present in gastric adenocarcinoma specimens,32 and nuclear accumulation of β-catenin is increased within gastric adenomas and foci of dysplasia,33–36 suggesting that aberrant activation of β-catenin precedes the development of gastric cancer. Since β-catenin is overexpressed within H. pylori-associated premalignant and malignant lesions, and regulates the transcription of genes that have been implicated in tumour initiation, it is likely that β-catenin is a central component in regulation of epithelial responses to H. pylori that may directly promote tumourigenesis.
Disruption of apical-junctional complexes is also linked to carcinogenesis and one group of tight junction constituents that mediate cell-cell adhesion are claudins. Claudins exert critical roles in regulating paracellular permeability and maintenance of cell polarity and aberrant function of specific claudins has been associated with cancer.14 Claudin-7 is localised to the apical tight junction and to the basolateral membrane of various tissues including intestinal epithelium.14 Genetic deficiency of claudin-7 in mice leads to increased intestinal cell proliferation and inflammation,17 and loss of claudin-7 is a negative prognostic factor for breast carcinoma and squamous cell carcinoma.14 ,37 The regulation of claudin-7 expression, however, is incompletely defined. One study demonstrated that recruitment of claudin-7 to tight junctions in mouse epithelial cells is regulated by EpCAM,38 while another report revealed that forced expression of snail leads to a reduction in claudin-7 expression.18 Our current findings extend these results by demonstrating that suppression of claudin-7 is also regulated by β-catenin, which is activated by CagA, and these results are mirrored in human gastric tissue harvested from H. pylori-infected subjects.
Another unique aspect of the current work is that it establishes 3D gastroids as a model that more closely recapitulates events occurring within the H. pylori-infected stomach.27 Our results demonstrate that gastroids have the ability to expand into single-layered epithelial spheroid structures that consist of mucus cells, parietal cells, G-cells, mucus neck cells, D-cells and ECL cells. This epithelial spheroid structure (gastroid) surrounds a sealed central lumen into which apoptotic cells are extruded, which recapitulates events occurring within gastric mucosa. Thus, gastroids can be described as having the capacity to develop into a self-organising differentiation axis. We successfully infected gastroids with H. pylori via microinjection, which resulted in mislocalisation of occludin at the tight junction, a response identical to what we previously demonstrated in H. pylori-infected MKN28 gastric epithelial cells and infected gastric epithelial cells in vivo.16 We also report increased snail expression in H. pylori-infected MKN28 gastric epithelial cells, gastric epithelium from human subjects, and gastroids. Snail can oscillate between the nucleus and the cytoplasm; however, previous reports have shown that snail is predominantly localised to the cytoplasm in human gastric adenocarcinoma specimens.39 In gastroids, similar to human specimens, we have shown that H. pylori increases expression of snail and that this increase is predominantly localised to the cytoplasm. Additionally, we have previously reported in gerbil models, as well as infected human subjects, that β-catenin is translocated to the nucleus in response to cag-positive H. pylori strains;8 ,13 these findings are consistent with our current results in gastroids where β-catenin is translocated to the nucleus following infection with cag-positive H. pylori. Overall, our findings indicate that H. pylori are effectively recognised by gastroids and that this system may be used as both a discovery model and to validate findings in human gastric mucosa.
Gastroids represent an important and novel model for studies of microbially induced disease. The majority of in vitro models that focus on H. pylori pathogenesis use multiple passaged cell lines, many of which contain mutations and are derived from cancer specimens. In vivo models of H. pylori infection most commonly use mice or gerbils; however, these models are expensive and can be time consuming to generate. Isolated gastric glands are an alternative model for studies of H. pylori-epithelial interactions22; however, there are limitations of this model, including a shorter lifespan (approximately 7 days) compared with gastroids that can survive for up to 9 months.27 Additionally, H. pylori does not induce proliferation in the isolated gastric gland model, which is different from the hyperproliferative response that develops within H. pylori-infected gastric mucosa and gastroids. Gastroids also effectively bridge in vitro and in vivo models by providing a culture system that can be readily generated from non-transformed gastric epithelium and gastroids contain the major cell types found within gastric glands. Although we have used gastroids to model H. pylori interactions with the gastric epithelium, this system could readily be adapted to study other host-microbial interactions. Further, we anticipate that this novel infection model will facilitate more in-depth research into the molecular mediators of gastric cancer that are initiated by H. pylori.
Using this novel system, we demonstrated that wild-type H. pylori, but not an isogenic cagA− mutant, significantly increases proliferation and that this is dependent on β-catenin signalling, supporting a model in which CagA-driven β-catenin activation leads to enhanced cell production. These findings are consistent with our previous data in both MKN28 gastric epithelial cells and a gerbil model demonstrating that H. pylori increases proliferation in a cag-dependent manner, which occurs via activation of β-catenin.13 The use of gastroids also revealed a new finding that claudin-7 expression was reduced in infected organoids, which guided mechanistic in vitro studies implicating β-catenin as a heretofore undescribed regulator of claudin-7. Of interest, a previous study using a different mouse model, hypergastrinaemic INS-GAS mice, demonstrated that chronic infection with another Helicobacter species, H felis, led to an increase in claudin-7 expression within gastric epithelium 6 months postchallenge.40 However, H felis lacks the cag PAI,41 emphasising the need to perform long-term studies in rodent models challenged with Helicobacter strains that harbour a functional cag island as a means to dissect the collective effects of H. pylori on claudin-7 expression in vivo.
In addition to host and bacterial constituents, environmental conditions can also modify the risk for H. pylori-induced carcinogenesis, for example, high salt diets, which have been linked epidemiologically to increased gastric cancer rates,42 accelerate the development of H. pylori-induced gastric cancer in rodents.43 Iron deficiency is also associated with an increased risk for gastric cancer.44 ,45 H. pylori infection contributes to iron deficiency,46 and iron depletion can augment the virulence potential of bacterial pathogens.47–50 We have demonstrated that CagA, which is required for β-catenin activation,8 ,9 ,13 facilitates H. pylori colonisation via iron acquisition from polarised epithelial cells51 and that iron depletion augments assembly of the cag type IV secretion system and cancer.52 Gastroids offer enormous potential as an innovative system to dissect these complex relationships as they can be subjected to different concentrations of dietary elements such as salt or iron, can be infected with wild-type or isogenic mutant H. pylori strains and can be generated from wild-type or genetically deficient mice.
In conclusion, we used a novel ex vivo 3D system to show that H. pylori induces proliferation and that this is dependent upon the H. pylori virulence factor CagA and β-catenin. Additionally, H. pylori alter the expression of claudin-7 in human gastric tissue and in gastric epithelial cells via signalling pathways that involve β-catenin and snail. Such investigations will not only improve our understanding of H. pylori-induced cancer, but will also provide mechanistic insights into other infection-related malignancies that arise within the context of inflammation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors: All authors included in this manuscript fulfil the criteria of authorship as follows: LEW: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, statistical analysis. MBP, RC, AD, JMN: acquisition of data. MS, EA, RF, DAI, YZ, MHM, NS, PC, KTW: material support, critical revision of manuscript for intellectual content. RMP: study concept and design, analysis and interpretation of data, drafting of the manuscript, critical revision of manuscript for important intellectual content, statistical analysis, obtained funding, study supervision.
-
Funding Supported in part by National Institutes of Health grants CA 153539 (JMN), AT 007324 (RC), CA 028842 (KTW, PC), DK 053620 (KTW), DK 58587, CA 77955, CA 116087 and DK 058404 (RMP).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Institutional Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.