Article Text

Abstract
Background and aims Gata6 is required to complete and maintain acinar differentiation in the mouse pancreas. Pancreas-specific Gata6 ablation during development causes extensive and persistent acinar-ductal metaplasia, which is considered an initial step of mutant KRas-driven carcinogenesis. Therefore, the Gata6-null pancreas might represent a tumour-prone environment. We investigated whether Gata6 plays a role during pancreatic tumorigenesis.
Design We analysed genetically engineered mouse models and human pancreatic ductal adenocarcinoma (PDAC) cell lines, using a combination of histopathological studies, genome-wide expression and chromatin immunoprecipitation experiments to understand the role of Gata6 in the initiation and progression of KRasG12V-driven tumours
Results We show that Gata6 maintains the acinar differentiation programme, both directly and indirectly, and it concomitantly suppresses ectopic programmes in the pancreas. Gata6 ablation renders acinar cells more sensitive to KRasG12V, thereby accelerating tumour development. Gata6 expression is spontaneously lost in a mouse model of KRasG12V-driven PDAC, in association with altered cell differentiation. Using a combination of ChIP-Seq and RNA-Seq, we show that Gata6 exerts its tumour-suppressive effect through the promotion of cell differentiation, the suppression of inflammatory pathways, and the direct repression of cancer-related pathways. Among them is the epidermal growth factor receptor (EGFR) pathway, the activity of which is upregulated in the normal and preneoplastic Gata6-null pancreas. Accordingly, GATA6-silencing in human PDAC cells leads to an upregulation of EGFR.
Conclusions We propose that, in the pancreas, Gata6 acts as a tumour suppressor by enforcing acinar cell differentiation, by directly and indirectly repressing ectopic differentiation programmes, and by regulating crucial cancer-related gene expression pathways.
- DIFFERENTIATION
- CARCINOGENESIS
- CELL BIOLOGY
- PANCREATIC CANCER
- MOLECULAR MECHANISMS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Gata6 is required for the maintenance of acinar differentiation in the mouse pancreas.
Pancreas-specific Gata6 knockout mice develop extensive and persistent acinar-ductal metaplasia (ADM).
ADM is an initial step of KRasG12D-driven pancreatic tumorigenesis in mice.
What are the new findings?
Gata6 ablation accelerates KRasG12V-driven tumorigenesis.
Gata6 is spontaneously lost in KRasG12V-driven murine pancreatic ductal adenocarcinoma (mPDAC), in association with altered differentiation.
Gata6 directly favours the acinar differentiation programme, represses non-pancreatic epithelial programmes, and regulates multiple pathways implicated in inflammation and cancer.
Gata6 regulates EGFR expression in mouse preneoplastic pancreas and in mouse and human PDAC.
Gata6 belongs to a new type of genes with context-dependent oncogenic and tumour suppressor effects.
How might it impact on clinical practice in the foreseeable future?
Expression levels of GATA6 might be predictive of prognosis and therapy response in patients with PDAC.
Results of clinical studies using EGFR inhibitors should be reanalysed in relationship with GATA6 genomics status and protein expression.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal disease with a 5-year overall survival of <5%.1 The most common alterations present in PDAC are activating mutations of the KRAS oncogene (>95% of cases) and inactivation of the tumour suppressors CDKN2A (>95%), TP53 (50–75%) and SMAD4 (55%).2 ,3 The additional genes involved in this tumour appear to play a role only in a small fraction of cases.3 ,4 KRAS mutations are considered as an initiating event because they are often detected in preneoplastic lesions (pancreatic intraepithelial neoplasias—PanINs) of the lowest grade as well as in tissues without PDAC.5 Pancreas-specific expression of mutant KRas in engineered mouse models is sufficient to initiate murine PDAC (mPDAC), although additional genetic alterations or induction of pancreatitis are required for tumour progression.6–10 Expression of mutant KRas in the acinar compartment induces transdifferentiation to a ductal-like phenotype through a process called ‘acinar-ductal metaplasia’ (ADM).7 ADM precedes PanIN formation in PDAC models,7 ,10–12 suggesting that it is an early event in mPDAC development. Persistent ADM is characteristic of chronic pancreatitis, a risk factor for PDAC;13 it is frequently associated with PanINs in patients with PDAC,14 and is therefore considered a preneoplastic lesion.15
We recently generated pancreas-specific Gata6 knockout mice, demonstrating that Gata6 is not essential for pancreas development after E10.5, but it is required for the complete differentiation and maintenance of the acinar compartment. GATA6 is a member of the GATA family of zinc-finger transcription factors sharing high homology and binding to the conserved WGATAR consensus sequence.16 Functional specificity of GATA proteins results mainly from divergent spatial and temporal expression patterns and from the interaction with cofactors. Gata4 and Gata6 are broadly co-expressed in endodermal tissues and share redundant functions during pancreas development, but they have distinct roles in the adult pancreas, where Gata4 cannot compensate for the loss of Gata6.17–19
In the normal pancreas, Gata6 is essential for the completion and maintenance of the acinar differentiation programme through the direct transcriptional regulation of genes coding for master acinar transcription factors, including Rbpjl and Mist1, as well as some digestive enzymes, such as pancreatic lipase (Pnlip). Gata6 ablation in multipotent progenitors resulted in quasi-normal histology in young adult mice but caused massive acinar atrophy, epithelial-adipocyte transdifferentiation, and extensive and persistent ADM in older mice, suggesting that a Gata6-null background might represent a tumour-prone environment.17
It has been proposed that a failure to complete acinar differentiation can sensitise cells to mutant KRas. Therefore, here we investigate the role of Gata6 in mPDAC and show that it has a tumour suppressive function. In the mouse pancreas, Gata6 supports a tissue-specific epithelial phenotype, represses ectopic non-pancreatic differentiation programmes, and participates in the control of inflammatory and cancer-related pathways, including Egfr and its downstream effectors. Its ablation favours KRasG12V-driven tumorigenesis, suggesting a tumour suppressor role. In agreement with this notion, Gata6 expression is spontaneously lost in a subset of mPDACs, in association with altered differentiation.
Material and methods
Mice
The following mouse strains were used: Gata6loxP/loxP;Ptf1a-Cre+/KI 17 and KRasLSLG12V-geo.8 For the analysis of Gata6 expression in mutant KRas-driven tumours, tissue samples were taken from >1-year-old KRas+/LSLG12V-geo;Ptf1a-Cre+/KI, KRas+/LSLG12V-geo;Elas-tTA/tetO-Cre that were not treated with doxycycline, and KRas+/LSLG12V-geo;Trp53lox/lox;Elas-tTA/tetO-Cre10 treated with doxycycline until 8 weeks to induce Cre activation in the adult. Mice were bred and maintained at the Spanish National Cancer Research Centre (CNIO) under sterile and pathogen-free conditions. All experiments were approved by the Animal Ethical Committee of Instituto de Salud Carlos III and performed in accordance with the guidelines for Ethical Conduct in the Care and Use of Animals as stated in The International Guiding Principles for Biomedical Research involving Animals, developed by the Council for International Organisations of Medical Sciences (CIOMS).
Histopathology and immunohistochemistry
Tissues were fixed in buffered formalin and embedded in paraffin. For histopathological analysis, pancreata were serially sectioned (3 μm) and every 10th section was stained with H&E. Areas of inflammation, ADM and murine PanINs (mPanINs) were measured with Pannoramic Viewer V.1.15 software. Quantification was performed with the Imagej software. Liver and lung samples were serially sectioned at 5 μm and every 20th section was stained with H&E and analysed for the presence of micrometastases. Immunostaining was performed using standard protocols.17
Gene expression analysis
Total RNA was extracted from total pancreas using guanidine thiocyanate (GTC) lysis followed by phenol chloroform extraction, and from cells using Trizol. qPCR was performed on cDNA using SYBR-green reagents.
Protein analysis
Total protein extracts were prepared in Laemmli buffer and sonicated. SDS-PAGE–western blotting was performed using standard protocols.
Statistical analysis
Data are provided as mean±SEM. Statistical analysis was done using two tailed Student t test or one-tailed Fisher's test and significance was considered for p<0.05. All statistical analyses were performed using VassarStat.net.
RNA-Seq and ChIP-Seq data were deposited in GEO under the following accession numbers: GSE47537 and GSE57090, respectively.
Online supplementary material contains extended experimental procedures and results, including Gata6P−/− vs Ctrl RNA-Seq analysis, GATA6 ChIP-Seq data, gene set enrichment analysis (GSEA) primers used in the study, and online supplementary figures.
Results
Gata6 ablation accelerates KRasG12V-driven pancreatic tumorigenesis
Increasing evidence suggests that master cell fate regulators are frequently lost in cancer in association with altered differentiation, leading to the activation of ectopic differentiation programmes, that is, metaplasia.20 Consistent with this notion, metaplastic changes—such as those observed in Gata6P−/−mice—might render cells more sensitive to pro-oncogenic events. To determine whether Gata6 can participate in PDAC development, we generated KRas+/LSLG12V-geo;Ptf1a-Cre+/KI;Gata6loxP/loxP (KRas*;Gata6P−/−) mice, where the mutant KRasG12V allele is activated—and Gata6 is concomitantly deleted—in Ptf1a+ pancreatic multipotent progenitors around day E9.5–10.8 ,17 ,21 In agreement with the highly efficient recombination induced by Ptf1a-driven Cre expression in the pancreas reported in other studies,8 ,17 ,21 we could not detect Gata6-expressing epithelial cells in the pancreas of KRas*;Gata6P−/− mice (see online supplementary figure S1). We analysed pancreatic histology at 5 and 25–28 weeks of age. At 5 weeks, control KRas+/LSLG12V-geo;Ptf1a-Cre+/KI mice (KRas*) showed small metaplastic foci and few low-grade mPanINs (figure 1A), whereas KRas*;Gata6P−/− mice showed diffuse oedema, large areas of inflammation with infiltrating macrophages (see online supplementary figure S2A), extremely abundant multifocal ADM (including areas of mucinous metaplasia and tubular complexes), and few low-grade mPanINs (figure 1A,B). The number of mPanINs/mouse was similar in both mouse strains (not shown). One 5-week-old KRas*;Gata6P−/− mouse, out of 8, developed a highly proliferative, large carcinoma (see online supplementary figure S2B), a finding that was never made in KRas* mice of that age. Stat3 activation has been implicated in KRas-induced pancreatic tumorigenesis.22 At 8 weeks, higher levels of p-Stat3 were detected in KRas*;Gata6P−/− mice compared with KRas* mice, supporting its involvement in the acceleration of tumour progression resulting from Gata6 loss. By contrast, p-Erk1/2 was unaffected (see online supplementary figure S2C).
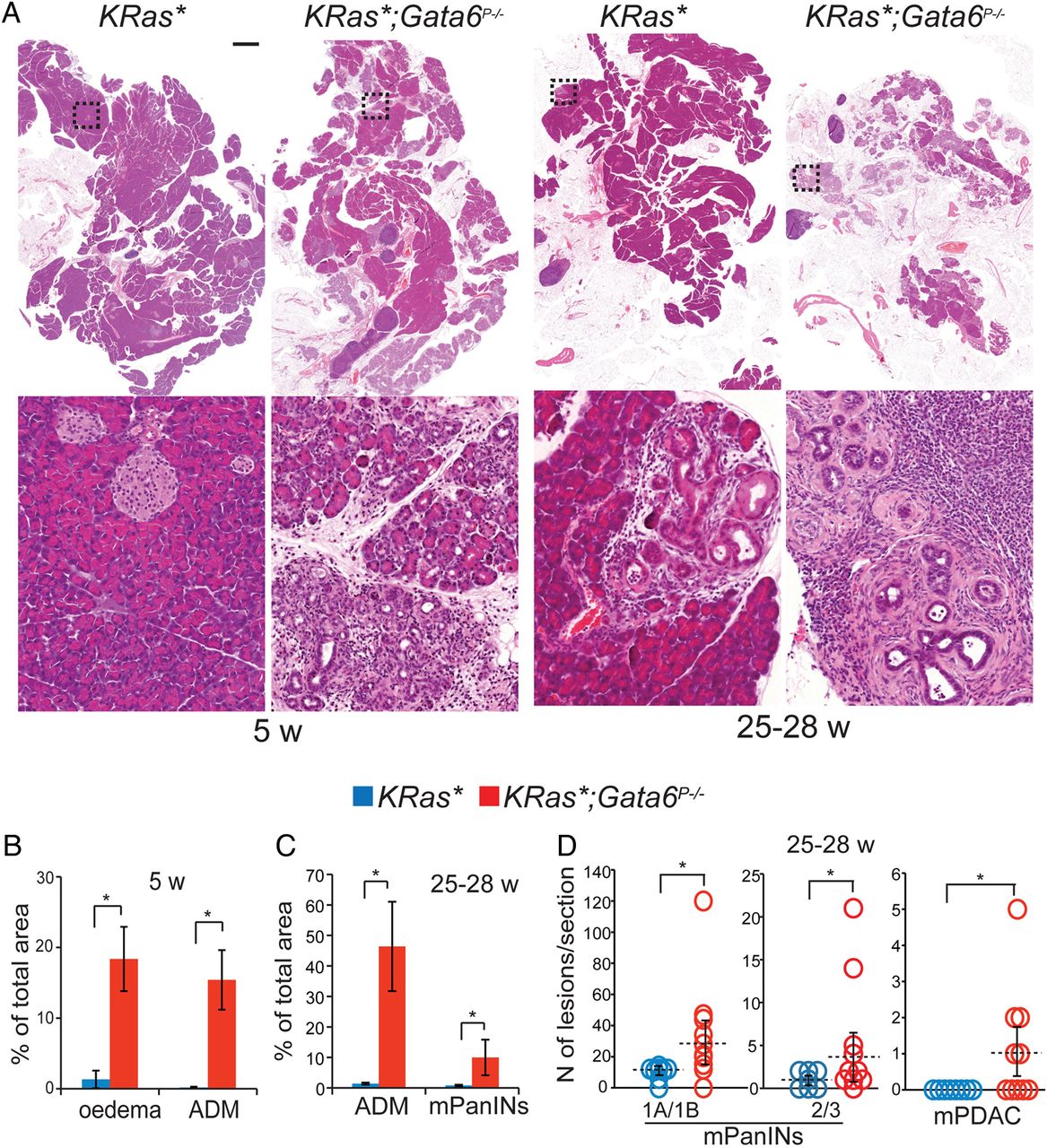
Gata6 suppresses KRasG12V-driven pancreatic tumorigenesis. (A) H&E staining of pancreatic sections from 5-week-old or 25–28-week-old KRas* and KRas*;Gata6P−/− mice. Higher magnification of the insets is shown under low-power images. Scale bar: 2 mm. (B) Quantification of oedema and acinar-ductal metaplasia (ADM) in 5-week-old KRas* (blue) and KRas*;Gata6P−/− (red) pancreata, measured as a percentage of total area. Data are mean±SEM KRas* (n=5); KRas*;Gata6P−/− (n=4) *p<0.05, **p<0.01. (C) Quantification of the area occupied by ADM and murine pancreatic intraepithelial neoplasias (mPanINs) in 25–28-week-old KRas* and KRas*;Gata6P−/− mice, represented as a percentage of total pancreas area. Data are mean±SEM (n=4 for both groups), *p<0.05. (D) Quantification of low-grade lesions (PanIN-1A and PanIN-1B (1A/1B in the graph)), high-grade lesions (mPanIN-2 and mPanIN-3 (2/3 in the graph)) and carcinomas (mPDAC) in 25–28-week-old KRas* (blue) and KRas*;Gata6P−/− (red) mice. KRas*(n=8), KRas*;Gata6P−/− (n=11), *p<0.05.
At 25–28 weeks, the number of mPanINs (both low-grade and high-grade) was significantly higher in KRas*;Gata6P−/− mice, as was the area of both mPanINs and ADM. At this age, carcinomas developed in 5/11 KRas*;Gata6P−/− mice and in 0/8 KRas* mice (p=0.035; figure 1C, D). All the carcinomas were scored as moderate/well differentiated.
We followed a cohort of 23 KRas* and 17 KRas*;Gata6P−/− mice until age 50–70 weeks. By that time, 16/17 (94.1%) KRas*;Gata6P−/− mice had developed large carcinomas while only 15/23 (65.2%) of the KRas* mice had detectable mPDAC (p=0.031). Importantly, 2/17 KRas*;Gata6P−/− mice had macroscopic liver metastases and one had peritoneal metastasis (not shown), whereas macroscopic metastases were never detected in KRas* mice (n=23). We further compared the presence of micrometastases using H&E-stained serial sections of livers and lungs from both mouse strains. We did not find significant differences in the occurrence of micrometastatic foci in KRas* vs KRas*;Gata6P−/− mice: liver 3/9 (33%) vs 4/12 (33%); lungs 3/10 (30%) vs 2/8 (25%) (not shown). These findings indicate that loss of Gata6 at the time of tumour initiation does not result in an increased rate of metastases, but it may affect their growth rate.
KRas*;Gata6P−/− mice also displayed acinar atrophy, as did Gata6P−/− mice. Of note, mPanINs or mPDAC were never observed in Gata6P−/− mice (not shown), even at age >2 years. Therefore, Gata6 loss is not sufficient for tumour initiation, but it favours KRasG12V-driven tumorigenesis.
Spontaneous Gata6 downregulation in KRas*-driven mPDAC is associated with altered differentiation
KRas* mice develop mPDAC around 1 year of age, recapitulating the human disease, which generally presents at an advanced age. We reasoned that KRas*-driven mPDAC must acquire additional genetic alterations23 that are spontaneously selected during tumour development. To determine whether Gata6 participates in the progression of tumours driven exclusively by mutant KRasG12V, we analysed its expression in the pancreas of >1-year-old KRas* or KRas+/LSLG12V-geo;Elas-tTA/tetO-Cre mice with mPDAC: Gata6 was consistently detected in mPanINs surrounding mPDACs (figure 2A, B-1), but it was completely or focally undetectable in 11/21 tumours (figure 2A), indicating that Gata6 loss of expression is selected during KRasG12V-driven tumorigenesis. Notably, Gata6 expression was also lost in 5/5 tumours from KRas+/LSLG12V-geo;Trp53lox/lox;Elas-tTA/tetO-Cre mice, where Cre expression was induced at 8 weeks.24

Gata6 expression is lost during KRasG12V-driven tumorigenesis, in association with poor differentiation. (A) Gata6 expression in Gata6neg and Gata6pos tumours from >1-year-old KRas* mice, detected by immunohistochemistry. Highlighted regions are shown in higher magnification in (B). Scale bars: 200 μm. (B) H&E staining and expression of Gata6 and E-cadherin in two regions of a Gata6neg mPDAC and in a region of a Gata6pos mPDAC, both taken from >1-year-old KRas* mice, detected by immunohistochemistry. Black arrowheads: acinar-ductal metaplasia, white arrowhead: low-grade murine pancreatic intraepithelial neoplasias (mPanIN). Scale bars: 100 μm.
The histopathological analysis of a subset of mPDAC revealed that 5/6 Gata6neg tumours and 1/8 Gata6pos tumours contained areas of moderate/poor differentiation (p=0.025). We also analysed the expression of pan-epithelial (E-cadherin) and ductal (Krt19) differentiation markers. Among KRasG12V-induced mPDAC, all Gata6pos tumours (n=10) expressed high levels of E-cadherin (figure 2B-3) and Krt19 (not shown). In contrast, areas of mPDAC with either broad or focal Gata6 losses (n=11) displayed reduced expression of E-cadherin and Krt19, indicating loss of the normal pancreatic epithelial differentiation programme (figure 2B-2). Similarly, 5/5 tumours from KRas*/p53P−/− mice expressed lower E-cadherin in the areas that were Gata6neg (not shown). Taken together, these observations demonstrate that Gata6 expression is selectively lost in a subset of KRasG12V-induced mPDAC featuring loss of cell differentiation, supporting a tumour suppressive function for Gata6.
Gata6 stabilises the pancreatic phenotype and suppresses non-pancreatic differentiation
To acquire a comprehensive understanding of the mechanisms through which Gata6 suppresses KRasG12V-driven tumorigenesis, we performed deep sequencing on RNA (RNA-Seq) from 6-week-old to 8-week-old Gata6lox/lox;Ptf1a-Cre+/KI (Gata6P−/−) mice and Ptf1a-Cre+/KI (Ctrl) controls. At this age, the pancreas of Gata6P−/− mice is histologically normal but the acinar transcriptional programme is already significantly altered.17 The RNA-Seq showed that 1826 genes were differentially regulated in Gata6P−/− mice with a p value <0.05; 1307 genes were downregulated and 518 were upregulated (see online supplementary table S1). We analysed 16 genes using quantitative RT-PCR (RT-qPCR) and confirmed the differential regulation of all of them (see online supplementary figure S3).
In addition to the reported downregulation of transcripts coding for the pancreas-specific components of the Pancreatic Transcription Factor 1 (PTF1) complex and digestive enzymes, the RNA-Seq revealed the de-repression of ectopic endodermal differentiation programmes. The activation of genes belonging to the gastric (Pgc, Muc6, Gkn3),25–27 intestinal (Vil1, Lgals4),28 ,29 liver (Alb, Serpina6),30 and lungs (Aqp5)31 differentiation programmes was confirmed by RT-qPCR (figure 3A). A similar de-repression of the liver differentiation programme has been reported for another important regulator of acinar differentiation, Rbpjl,32 suggesting that this mechanism might be common among transcription factors controlling cell identity.
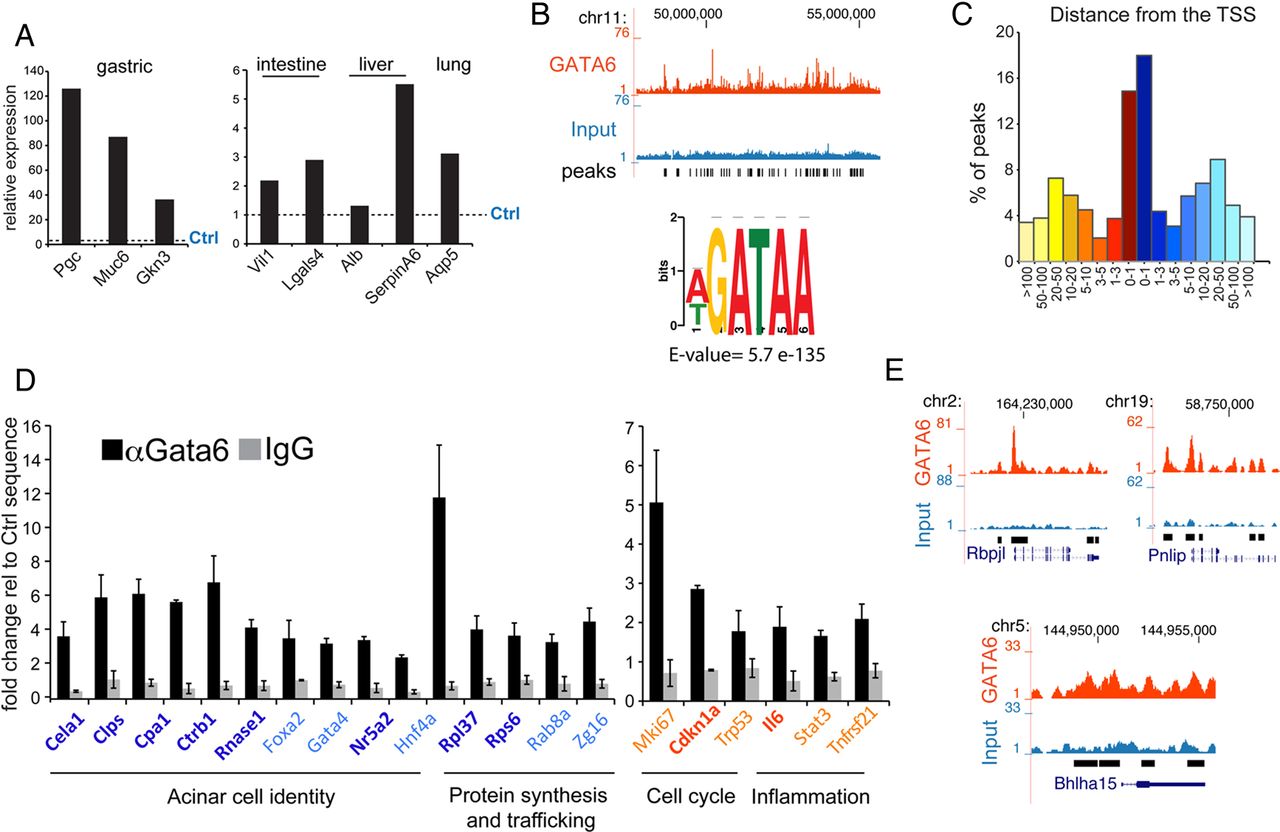
Gata6 directly regulates differentiation, inflammation and cancer-related pathways. (A) Expression of non-pancreatic genes in the pancreas of 6–8-week-old Gata6P−/− mice (n=4), measured by RNA-Seq, relative to Ctrl mice (dotted line). (B) Top: Representative genomic distribution of Gata6 in a region of mouse chromosome 11, as displayed in the UCSC genome browser. Tag counts (top) (red, Gata6-ChIP; blue, input) and called peak (bottom) tracks are shown. Bottom: core matrix obtained for the most significantly enriched motif found in the Gata6 ChIP-Seq peaks. (C) Distribution of Gata6 ChIP-Seq peaks according to their distance from the closest transcription start site (TSS). (D) Gata6 binding to the promoter of the indicated genes detected by ChIP-qPCR in total mouse pancreas. Colours represent the regulation of target genes in previously reported RT-qPCR or in RNA-Seq: dark blue=statistically significant downregulation; light blue=downregulated, not significant; red=upregulated, significant; orange=upregulated, not significant; black=unchanged. Data are presented as mean±SEM of >3 independent experiments. ChIP-qPCR data are represented as % of input, normalised against a control sequence, compared to non-specific IgG binding. Enrichment for Gata6 was significant (p<0.05) at all locations. (E) Representation of the ChIP-Seq peaks found in the promoter of the indicated genes, as displayed in the UCSC genome browser. Tag count (red, Gata6-ChIP; blue, input), called peaks, chromosome coordinates and RefSeq gene tracks are shown.
Gata6 directly regulates differentiation, inflammation and cancer-related pathways
To identify the genetic programme(s) directly controlled by Gata6, we performed chromatin immunoprecipitation followed by massive parallel sequencing (ChIP-Seq) using total pancreatic extracts from 5-week-old to 14-week-old Ctrl mice. A total of 7733 Gata6 binding sites were identified with a cut-off of false discovery rate (FDR)<1% (see online supplementary table S2). The canonical DNA consensus sequence for GATA factors was the most enriched motif in the sequenced tags, as assessed with the MEME ChIP suite (E value=5.7e-135, figure 3B). Gata6 peaks were preferentially found (32%) within 1 kb from the transcriptional start site (TSS) of coding genes (figure 3C).
We then performed an intersection of the results of Gata6-binding, identified by ChIP-Seq, and RNA-Seq expression changes in the pancreas of 6–8-week-old Gata6P−/− vs Ctrl mice. Of 1307 genes whose mRNAs were significantly downregulated in Gata6P−/− mice, 379 (29%) had a Gata6 peak, and 249 of them (19% of all) had a Gata6 peak within 3 kb from the TSS. Of 518 genes whose mRNAs were significantly upregulated in Gata6P−/− mice, 77 (14.9%) had a Gata6 peak, and 34 of them (6.6% of all) had a Gata6 peak within 3 kb from the TSS. A significantly higher fraction of the genes that were downregulated in Gata6P−/− mice appear under the direct control of Gata6, compared with those that were upregulated (p<0.001), suggesting that Gata6 mainly acts as a transcriptional activator.
Gata6 controls broadly acinar function
GSEA of the genes included in the overlap revealed pathways that were significantly downregulated in Gata6P−/− mice, including those involved in protein biosynthesis and mitochondrial metabolism (table 1, see online supplementary table S3). The transcripts of multiple genes coding for proteins of the large and small ribosomal subunits (ie, Rpl37, Rps6, Rab8), as well as those coding for components of the traffic and secretion machinery (ie, Zg16, Rab8, Srp9, Srp14, Srp19), were downregulated. ChIP-qPCR confirmed the binding of Gata6 at the proximal promoters of several of them (figure 3D), indicating that Gata6 regulates directly and broadly the protein-secretion function of acinar cells.
Gene sets enriched in RNA-Seq and in the RNA-Seq/ChIP-Seq integration
GSEA does not contain a ‘pancreatic acinar’ gene set. Therefore, we assessed the acinar transcriptional programme manually and confirmed by ChIP-qPCR that Gata6 binds the proximal promoter of genes belonging to this programme.17 Expanding our previous observations, we show that Gata6 is a direct regulator of genes coding for both acinar master transcription factors (Rbpjl and Mist1—also known as Bhlha15), as well as digestive enzymes whose transcripts are downregulated in Gata6P−/− mice (Pnlip, Amy2a5, Cela1, Clps, Cpa1, Ctrb1, Pnliprp1, Rnase1) (figure 3D, E). Additional transcription factors participating in the acinar programme had a Gata6 peak and showed a trend towards lower mRNA levels in Gata6P−/− mice (Foxa2, Gata4, Nr5a2, Hnf4a), although the differences did not always reach statistical significance (figure 3D, see online supplementary table S1).
Gata6 represses a pro-inflammatory programme in pancreatic epithelial cells
Pathways related to inflammation were over-represented among the genes with a Gata6 peak that were upregulated in the Gata6P−/− mice, although only two pathways appeared as significantly enriched due to the low number of genes (77) included in the analysis (table 1, see online supplementary table S3). GSEA performed on all genes overexpressed in Gata6P−/− mice confirmed that inflammation-related pathways were strongly induced (table 1, see online supplementary table S3). Gata6 binding to the proximal promoter of selected inflammatory genes (Il6, Stat3, Tnfrsf21) was confirmed by ChIP-qPCR (figure 3D). As aforementioned, at this age the pancreas of Gata6P−/− mice is histologically normal except for the presence of occasional CD45+ inflammatory cells (see online supplementary figure S4A). In order to exclude that these cells are responsible for the pro-inflammatory programme described above, we isolated acinar cells from wild-type and Gata6P−/− mice and cultured them shortly in suspension.33 We assessed by RT-qPCR the expression of Gata6, acinar-specific genes (Ptf1a, Cel, Cpa), markers of T lymphocytes (CD3d and CD3g), and several genes involved in inflammation (Ccl5, Ccl6, Ccl7, Ccl9, Cxcl14). At the time of cell isolation (day 1), we confirmed that Gata6 was efficiently knocked out (see online supplementary figure S4A). At this time point, we also observed that the levels of acinar-specific mRNAs, such as Ptf1a, Cel and Cpa, were reduced in Gata6P−/− cultures (see online supplementary figure S4B).17 At day 1, few inflammatory cells were present, as indicated by the detection of the T-lymphocyte markers CD3d and CD3g. However, by day 3, CD3d and CD3g transcripts were undetectable (see online supplementary figure S4C), while acinar cells from Gata6P−/− mice showed significantly increased expression of inflammation-related genes. Similar results were obtained at day 5 (see online supplementary figure S4D), indicating that the pro-inflammatory gene expression phenotype identified in the RNA-Seq experiment is not due to the influx of leucocytes in the pancreas. These results confirm that the pro-inflammatory programme controlled by Gata6 is epithelial cell autonomous.
Gata6 controls cancer-related pathways, including Egfr
The statistical cut-off used in the analysis of the RNA-Seq data was necessarily restrictive in order to avoid false positives. However, we reasoned that subtle but consistent changes in the expression of many genes involved in the same pathway might result in a global regulation of the entire programme. We then performed pathway analysis on all genes with a Gata6 peak (see online supplementary table S2). In addition to the pathways related to inflammation, those related to cell cycle were significantly over-represented (table 2, see online supplementary table S3). We confirmed Gata6 binding to the proximal promoter of cell cycle-related genes such as Mki67, Cdkn1a and Trp53 by ChIP-qPCR (figure 3D). Most of the genes with a Gata6 peak showed a trend towards differential regulation in Gata6P−/− mice in the RNA-Seq experiment, although the differences did not always reach statistical significance (see online supplementary table S1).
Gene sets enriched in ChIP-Seq
We then restricted the GSEA analysis to the genes with a Gata6 peak ±3 kb from the TSS. The ERBB-pathway was highly over-represented among Gata6 direct targets (table 3, see online supplementary table S3). EGFR has been shown to be essential for mutant KRas-driven PDAC development.34–36 Gata6 peaks were present at the Egfr locus, with a small peak right on the TSS and three additional peaks in the first intron (figure 4A). ChIP-qPCR confirmed Gata6 binding in the first intron (figure 4B) and Egfr mRNA was moderately upregulated in Gata6P−/− mice (log2 Fc=0.67). We analysed EGFR protein expression in the pancreas using immunohistochemistry. In comparison with controls, EGFR was focally overexpressed in 5-week-old Gata6P−/− mice, with a membrane-bound pattern (figure 4C, D). A strong and broader EGFR membrane expression was observed in KRas* mice within ADM and mPanIN areas, but the highest expression was observed in KRas*;Gata6P−/− mice, where diffuse and intense membrane and cytoplasmic EGFR staining was observed (figure 4C, D). These findings indicate that mutant KRas cooperates with Gata6 inactivation to upregulate EGFR. Interestingly, Egf mRNA was significantly downregulated in Gata6P−/− mice (log2 Fc=−2.07) and a Gata6 peak was observed in its proximal promoter, suggesting a direct regulatory feedback loop. Among the other ERBB ligands, Hbegf, Ereg, Areg, Epgn, Nrg1, Nrg2 and Nrg3 were not detectable in the RNA-Seq experiment, while Tgfa, Btc and Nrg4 were not significantly regulated (not shown).
Gene sets enriched in selected ChIP-Seq peaks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gata6 inhibits Egfr transcription in mouse and human cells. (A) Representation of the Gata6 peaks along Egfr. (B) ChIP-qPCR showing Gata6 binding to the first intron of Egfr—red arrow—as compared with an unrelated genomic region (Neg) and with non-specific IgG. Data are presented as mean±SEM of the absolute % of input obtained in three independent experiments; *p<0.05. (C) Quantification of red-green-blue (RGB) intensity corresponding to the expression of EGFR in the microphotographs shown in (D). Data are presented as mean±SEM (n=3 in each group); *p<0.05. (D) H&E and immunohistochemistry staining for total EGFR in the pancreas of 5–8-week-old mice of the indicated genotypes. Insets show higher magnifications of the highlighted area. (E) Expression of EGFR in GATA6-silenced PaTu8988S cells, measured by RT-qPCR. Two different shRNA constructs were used. Data are presented as mean±SEM of three independent experiments; *p<0.05. (F) Expression of EGFR, Phospho-EGFR and its activated downstream targets p-AKT and p-ERK1/2 in GATA6-silenced PaTu8988S, detected by western blot. GAPDH was used as loading control. (G) Scatter plot showing the correlation between GATA6 and EGFR expression in the joint analysis of four PDAC gene expression data sets.
The fibroblast growth factor receptor (FGFR) and platelet-derived growth factor (PDGF) pathways were also over-represented among Gata6 targets (table 3, see online supplementary table S3). Gata6 peaks were present within 3 kb from the TSS of multiple genes involved in both pathways (Fgfr1, Fgfr3, Fgf2, Pdgfrb, Pdgfa, see online supplementary table S2). Additionally, mRNA levels of Pdgfrb and Pdgfc were upregulated (log2 Fc=0.98 and 1.59, respectively) and the mRNA level of Fgf21 was downregulated (log2 Fc=−1.97) in Gata6P−/− mice; other genes belonging to these pathways showed some trend to differential regulation. In conclusion, Gata6P−/− mice displayed extensive deregulation of growth signalling pathways.
GATA6 regulates EGFR in human PDAC
PDAC cells with low levels of GATA6 and a quasi-mesenchymal gene expression signature have been reported to be more resistant to EGFR inhibitors.37 To elucidate whether GATA6 directly regulates EGFR in human PDAC, we knocked it down in PaTu8988S cells expressing high levels of GATA6 with two different shRNA lentiviral constructs. Efficient knockdown was confirmed at the mRNA (not shown) and at the protein levels (figure 4F). EGFR mRNA was upregulated in GATA6-silenced PaTu8988S cells (shG6-1: 2.33-fold, p=0.025; shG6-2: 2.11-fold, p=0.048; figure 4E). Upregulation of total EGFR and p-EGFR, and activation of downstream effector pathways (p-ERK1/2 and p-AKT), were observed on GATA6 knockdown (figure 4F). Consistently, a moderate negative correlation between GATA6 and EGFR mRNA expression levels (r=−0.32, p<0.001) was observed in a meta-data set of gene expression in primary PDAC samples including four published data sets (figure 4G).38–41 We therefore uncover a novel transcriptional GATA6-dependent regulatory mechanism for EGFR that might be of therapeutic relevance.
Discussion
We identify Gata6 as a crucial regulator of epithelial and pancreatic cell differentiation endowed with tumour suppressive capacity. Its loss during tumorigenesis is associated with altered differentiation, strengthening the notion that genetic factors impinging on normal differentiation favour oncogenesis.
KRasG12V causes mPanINs and mPDAC formation when expressed from the endogenous promoter in embryonic pancreatic cells but not in adult, well-differentiated, acinar cells.10 Induction of a chronic pancreatitis-like lesion is sufficient to restore the permissiveness of adult acinar cells to oncogenic transformation, probably through the transient activation of a less mature phenotype.24 These observations suggest that incomplete acinar differentiation sensitises cells to the oncogenic effects of mutant KRas.12 We reported recently that Gata6 is dispensable for pancreas formation and that Gata6-null pancreata display a normal histology but fail to undergo complete differentiation. On the basis of these findings, we explored whether Gata6 could play a tumour suppressive role in KRasG12V-driven mPDAC and the mechanisms involved therein.
We find that inactivation of Gata6 in pancreatic progenitors—at the same time that KRasG12V is expressed—increases the number of preneoplastic lesions, including ADM, and accelerates tumour progression. However, the spontaneous loss of GATA6 expression in KRasG12V-induced tumours is most likely a late event in pancreatic carcinogenesis, since expression is maintained in mPanINs. This sequence is not fully recapitulated in the KRas*;Gata6P−/− model that we described here; the recently published dual recombinase system42 will allow elucidation of the effect of the late loss of GATA6 expression. Several additional observations further support the notion that Gata6 can act as a pancreatic tumour suppressor. Gata6 is spontaneously lost in KRasG12V-driven tumours and it is a target of the sleeping-beauty transposable element in an insertional mutagenesis screening performed in KRasLSL-G12D+/KI;Pdx1-Cre+/KI mice.23 Furthermore, GATA6 is lost in human PDAC in association with loss of pancreatic epithelial differentiation markers—such as E-cadherin—and GATA6 knockdown in cultured human PDAC cells leads to loss of canonical epithelial differentiation (Martinelli et al, unpublished). The fact that Gata6neg KRasG12V-driven tumours also showed a moderate/poor differentiation and low E-cadherin expression strengthens the notion that Gata6 plays similar roles in human and mouse PDAC. Finally, inhibition of Gata6 is essential for the tumour-promoting effect of nicotine in a mouse model of mutant KRas-driven pancreatic tumorigenesis.43
We have taken a genome-wide approach to further our understanding of how Gata6 inactivation/loss contributes to PDAC. We confirm that Gata6 is essential for complete differentiation of acinar cells both through the direct regulation of the genes coding for digestive enzymes and, indirectly, because transcription factors required for complete acinar differentiation, such as Mist1 and Nr5a2,11 ,44–46 are also Gata6 targets (see online supplementary table S2). In addition, we have unveiled that Gata6 controls unexpected aspects of acinar cell function including the regulation of the protein synthesis machinery—through the control of ribosomal proteins—and of protein secretion, thus orchestrating the full secretory differentiation programme. Interestingly, this is coupled with the repression of alternative cell lineages such that Gata6-null pancreata display features of altered epithelial identity.
These findings add to the increasing evidence that tissue-specific differentiation programmes are aberrantly regulated during tumour development and that de-differentiation is not only a hallmark but also a precursor of malignancy. This can result from the loss of master regulators of cell fate, such as Nkx2-1 in lung cancer, the loss of which leads to the de-repression of a latent gastric differentiation programme contributing to shape abnormal tumour phenotypes.20 As Nkx2-1, Gata6 represses non-pancreatic epithelial genes whose expression is abnormally detected in Gata6neg cells. Rbpjl—a PFT1 complex component and a direct Gata6 target—was shown to suppress the hepatic phenotype in acinar cells.32 The non-pancreatic epithelial genes that are expressed on Gata6 loss belong to several endoderm-specific differentiation programmes, mostly foregut specific, which most likely is related to the embryological origin of the pancreas.47 Foregut-specific transcripts can also be detected in low-grade PanINs, indicating that activation of non-pancreatic genes is an early event during tumour development.48 Gata6 was generally expressed in mPanINs, suggesting that other factors contribute to regulate the expression of non-pancreatic genes.
Beyond the control of cell differentiation, our analysis uncovers new functions, embedded in the Gata6-driven transcriptional programme, involved in cancer progression, namely cell cycle and inflammation, which can also contribute to its tumour suppressive effect. Gata6-null acinar cells display higher proliferation17 and now we provide evidence suggesting that this might be the consequence of Gata6 directly regulating the expression of genes involved in the cell cycle control. Furthermore, a detailed examination of the pancreas of young Gata6P−/− mice revealed occasional infiltrating macrophages, B and T lymphocytes (not shown). Our GSEA analyses uncovered a broad activation of inflammation-related pathways in Gata6-null pancreata, suggesting that Gata6 loss induces a pre-inflammatory environment in the pancreas, which might be exacerbated by mutant KRas, thereby accelerating tumour development. Importantly, the Gata6-dependent inhibition of the pro-inflammatory programme described is epithelial cell-autonomous, supporting the notion that epithelial-specific transcription factors can have a role in the control of inflammatory processes.
Collisson and colleagues have proposed that PDACs can be classified into three distinct subtypes (epithelial, quasi-mesenchymal and exocrine-like) differing in prognosis and, most importantly, response to therapy.37 In particular, PDAC cells with low GATA6 and a mesenchymal-like gene expression profile were more resistant to EGFR inhibitors. We show that GATA6 directly represses EGFR in normal and preneoplastic mouse pancreatic cells, as well as in human PDAC cells, and that the pathways downstream of EGFR are broadly activated when GATA6 levels are reduced. Among the drugs tested in combination with gemcitabine in the treatment of PDAC, only erlotinib and nab-paclitaxel have shown a significant impact on survival.49 ,50 The effect of the EGFR inhibitor erlotinib was small, possibly because only a fraction of patients benefit from the drug.49 Therefore, the identification of markers predictive of response is important to improve patient stratification. Since GATA6 broadly regulates epithelial differentiation, tumour subtypes differing according to GATA6 expression levels may display distinct sensitivity to other therapies against PDAC.
Prior studies have reported copy number gains/amplification of the GATA6 locus in a subset of PDACs,51–53 leading to the proposal that it is an oncogene, but the biological significance of the GATA6 gains/amplifications remains unclear. GATA6 has been proposed to regulate proliferation 52–54 and our data indicate that it binds to the promoter of cell cycle-related genes in the mouse pancreas. Different genetic contexts likely select for gains or losses of GATA6 in PDAC cells. In support of this notion, Gata6 is required for the tumorigenic effect of Apc inactivation in the mouse colon,55 strongly suggesting that it belongs to a novel type of cancer genes that can both promote and suppress tumours.
The previous studies in PDAC were at odds with the finding, in two independent reports, that high levels of GATA6 are associated with significantly better prognosis.37 ,54 Our work provides an explanation to this conundrum by showing that GATA6 has tumour suppressive functions. We propose that GATA6 can act as a tumour suppressor in the pancreas by locking epithelial cells in a differentiated state, thereby repressing multiple hallmarks of cancer. The study of a large series of patients with detailed genomic and clinical annotation (ie, those in the International Cancer Genome Consortium and The Cancer Genome Atlas) should contribute to unravelling the contrasting roles of GATA6 in human PDAC.
Acknowledgments
The authors thank M Barbacid, E Batlle, A Cano, O Domínguez, A Muñoz, O Graña, M Serrano and E Wagner for valuable contributions, Y Cecilia for animal handling, CNIO core facilities for continued support, and investigators who provided reagents or mice as indicated in the text.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online tables
Footnotes
Contributors PM made study concept and design, acquisition of the data, analysis and interpretation of the data, and drafting of the manuscript; FM made acquisition of the data, analysis and interpretation of the data; MC and EC-dSP made analysis and interpretation of the data; NdP made technical support; CG made acquisition and interpretation of the data and provided reagents; FXR made study concept and design, interpretation of the data, drafting of the manuscript, study supervision, and obtained funding. All authors read and commented on the final manuscript draft.
Funding This work was supported, in part, by grants SAF2007-60860, SAF2011-29530, and ONCOBIO Consolider from Ministerio de Economía y Competitividad (Madrid, Spain), RTICC from Instituto de Salud Carlos III, and grants 256974 and 289737 from the European Union Seventh Framework Programme to FXR. PM was recipient of a Juan de la Cierva grant from the Spanish Ministry of Science and Innovation. FM is the recipient of a grant from the International La Caixa PhD programme.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The supplementary tables contain all relevant material to be shared. The next generation sequencing data will be deposited in public databases. Any unpublished work or methodological details will be shared with the scientific community, on request.