Article Text

Abstract
Objective Hepatocellular carcinoma (HCC) is an aggressive malignancy with limited effective treatment options. An alternative strategy is to target cells, such as tumour-infiltrating macrophages, in the HCC tumour microenvironment. The CCL2/CCR2 axis is required for recruitment of monocytes/macrophages and is implicated in various aspects of liver pathology, including HCC. We investigated the feasibility of CCL2/CCR2 as a therapeutic target against HCC.
Design CCL2 expression was analysed in two independent HCC cohorts. Growth of three murine HCC cells was evaluated in an orthotopic model, a postsurgical recurrence model and a subcutaneous model in mice after blocking CCL2/CCR2 axis by a novel CCR2 antagonist or knocking out of host CCR2. In vivo macrophage or T cell depletion and in vitro cell coculture were further conducted to investigate CCL2/CCR2-mediated crosstalk between tumour-associated macrophages (TAMs) and tumour cells.
Result CCL2 is overexpressed in human liver cancers and is prognostic for patients with HCC. Blockade of CCL2/CCR2 signalling with knockout of CCR2 or with a CCR2 antagonist inhibits malignant growth and metastasis, reduces postsurgical recurrence, and enhances survival. Further, therapeutic blocking of the CCL2/CCR2 axis inhibits the recruitment of inflammatory monocytes, infiltration and M2-polarisation of TAMs, resulting in reversal of the immunosuppression status of the tumour microenvironment and activation of an antitumorous CD8+ T cell response.
Conclusions In patients with liver cancer, CCL2 is highly expressed and is a prognostic factor. Blockade of CCL2/CCR2 signalling suppresses murine liver tumour growth via activating T cell antitumour immune response. The results demonstrate the translational potential of CCL2/CCR2 blockade for treatment of HCCs.
- MACROPHAGES
- CELL BIOLOGY
- CHEMOKINES
- HEPATOCELLULAR CARCINOMA
- IMMUNOLOGY IN HEPATOLOGY
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Hepatocellular carcinoma (HCC) is an aggressive malignancy with limited effective treatment options. Targeting tumour-infiltrating macrophages is a promising strategy.
The CCL2/CCR2 axis is required for recruitment of monocytes/macrophages and is implicated in various aspects of liver pathology, including acute liver injury, chronic hepatitis, cirrhosis and tumorigenesis.
The function of the CCL2/CCR2 axis in HCC tumours and its potential as a therapeutic target for liver cancer remain elusive.
What are the new findings?
CCL2 is overexpressed in human HCCs and is an independent prognostic factor for patients.
The growth of CCL2-overexpressing liver tumour cells is dependent on host CCR2 in vivo.
Blocking CCL2/CCR2 with a CCR2 antagonist suppresses liver tumour growth and postsurgical recurrence in a CD8 T cell dependent manner.
Blockade of CCL2/CCR2 abolishes the cross-talk between tumour cells and macrophages and suppresses M2 macrophage polarisation and its immunosuppression of lymphocytes.
How might it impact on clinical practice in the foreseeable future?
Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling show promising potential for tumour-associated macrophages-based strategies for HCC treatment.
Introduction
Hepatocellular carcinoma (HCC) is a highly aggressive primary liver malignancy and the third leading cause of cancer-related death worldwide.1 ,2 Although surgical resection remains the most effective treatment for patients with early disease, nonsurgical approaches, such as chemotherapy, are necessary for patients, which are not suitable for hepatic resection.3 Nevertheless, large proportions of patients with HCC develop postsurgical recurrence and have a low response to chemotherapeutic agents, with a 5-year survival rate of only 30–40%. Therefore, there is a need to identify therapeutic strategies that circumvent these limitations.
In HCCs, the stromal component of tumours consists of fibroblasts, endothelial cells and tumour-infiltrating inflammatory cells.4 These cells, which produce a microenvironment that modifies the neoplastic properties of the tumour cells, are involved in tumour development, tumour control and response to treatment.5–7 Among inflammatory cells, tumour-associated macrophages (TAMs) are believed to promote cancer initiation and malignant progression.7–10 For example, there is an association between poor survival and increased macrophage density in lung cancer, thyroid cancer, prostate cancer and HCC.8 ,9 ,11 ,12 Under pathological conditions, Ly6Chigh inflammatory monocytes13—but not the Ly6Clow non-inflammatory subset—depend on the chemokine/chemokine-receptor CCL2/CCR2 pair for localisation to the site of inflammation, and subsequently give rise to classical macrophages and promote inflammatory diseases, including malignancies.14–16
Chemokines are a family of small, secreted proteins that have pleiotropic roles in inflammation-related pathological diseases, including cancer.17 Chemokine CCL2 is a multifunctional factor involved in various aspects of liver pathogenesis, including acute liver injury, chronic HBV/HCV infection, cirrhosis and tumorigenesis.18–20 CCL2 is secreted by hepatic stellate cells, hepatocytes, Kupffer cells and biliary epithelial cells.18 CCR2, the only known receptor for CCL2, is expressed on monocytes and macrophages within the liver. When CCL2 and CCR2 are upregulated in the liver, they promote macrophage accumulation, inflammation, fibrosis and steatosis.18 CCL2 also modulates macrophage polarisation by granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF.21 Further, the CCL2/CCR2 axis influences cell growth, angiogenesis, invasion and metastasis.22 ,23 In some preclinical cancer models, targeting of CCL2 is an effective therapeutic approach, and neutralising antibody against CCL2 has entered clinical trials for treatment of prostate cancer.24 ,25 However, the function of the CCL2/CCR2 axis in HCC tumours and its potential as a therapeutic target for liver cancer remain poorly understood.
The present investigation, involving use of three murine HCC tumour models and two independent, retrospective cohorts of patients with HCC, was designed to determine the role of the CCL2/CCR2 axis in HCC tumour growth and progression, to evaluate CCL2/CCR2 targeting for antitumour activity and to investigate the mechanism of action.
Materials and methods
Patient cohorts
In Cohort I, HCC tissue microarray chips containing 88 pairs of tumours and matched adjacent tissues were obtained from the Shanghai Biochip Company. The clinicopathological and follow-up data of patients were prospectively collected. In Cohort II, clinical samples were obtained from 96 patients with HCC treated at the Shanghai Eastern Hepatobiliary Surgery Hospital from January 2012 to February 2014.
Animal models
To establish the subcutaneous HCC model, HCC cells were injected into the right flanks of recipient mice. To establish an orthotopic HCC model, luciferase-labeled Hepa1-6 cells were orthotopically implanted into the livers of recipient mice. The establishment and growth of tumours were monitored by bioluminescent imaging with the Xenogen in vivo imaging system (IVIS). To construct the orthotopic recurrent model, mice bearing Hepa1-6 orthotopic tumours were anaesthetised for tumour resection. Mice with successfully removed primary tumours were determined by luciferase expression. Postsurgical recurrence of tumours was detected by bioluminescent imaging.
In vivo depletion of macrophages and CD8 cytotoxic lymphocytes
For macrophage depletion, Hepa1-6 tumour bearing mice were injected with 30mg/kg of gadolinium chloride (GdCl3) through the caudal vein, followed by repeated injections every 4th day to prevent repopulation of macrophages. For CD8 cytotoxic lymphocytes depletion, Hepa1-6 tumour bearing mice were intraperitoneally injected with anti-CD8 neutralised antibody, followed by repeated injection of CD8 antibody every 5th day.
Generation of tumour cell-educated macrophages
For tumour ‘education’, the conditioned medium (CM) was collected from murine Hepa1-6 liver cancer cells incubated in serum-free medium for 48 h and added to bone-marrow derived macrophages (BMDMs) for 24–48 h, after which the expressions of inflammatory cytokines and M1 and M2 macrophage markers were assayed.
Statistical analysis
All values were recorded as the means±SEM from at least two or three independent experiments. For the cell culture experiments, statistical significance was determined through a two-tailed Student's t test. Kaplan-Meier analysis and log-rank test were used to illustrate differences in patients' overall survival. The χ2 test was used to test for relationships between categorical variables. p Values<0.05 were considered statistically significant.
Results
In patients with liver cancer, CCL2 is highly expressed and is an independent prognostic factor
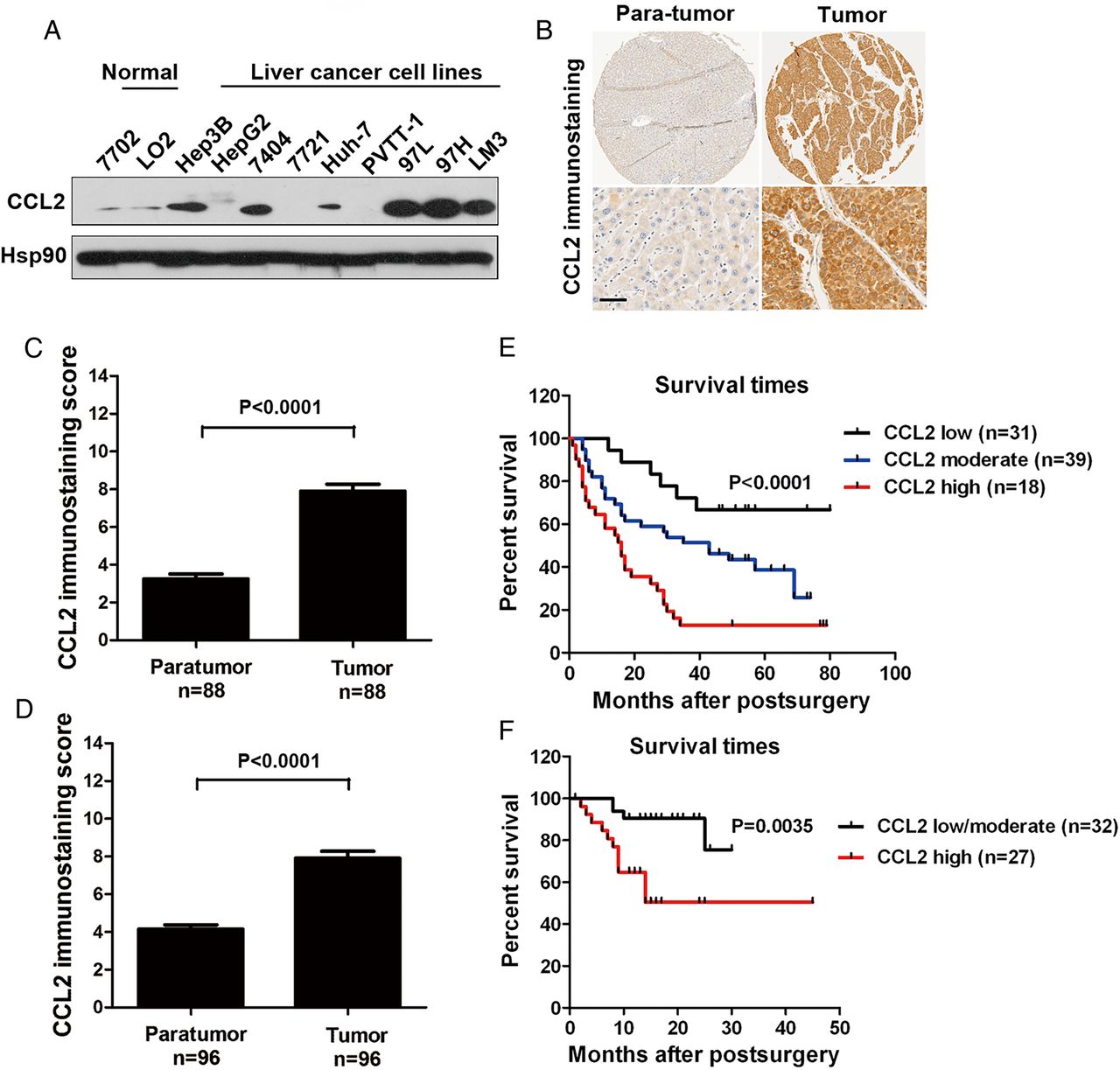
The protein expression levels of CCL2 in six of nine HCC cell lines were higher relative to levels in non-transformed hepatic cell lines (figure 1A). To confirm this finding, immunohistochemical expression of CCL2 was evaluated in tissue microarrays with tumours from two independent, retrospective cohorts of patients with HCC. In both cohorts, the amount of CCL2 protein was elevated in HCC tissues relative to their paired, adjacent, non-tumour tissues (figure 1B). Compared with the para-tumour tissues, 86.4% (76 out of 88 cases) and 74.0% (71 out of 96 cases) of HCC tumour tissues in Cohort I and Cohort II, respectively, showed increased expression of CCL2 (figure 1C, D and see online supplementary figure S1).

CCL2 is highly expressed in human liver cancers, and its levels correlate with patient survival. (A) By western blots, the expression of CCL2 was analysed in two normal human liver cell lines (HL-7702 and LO2) and nine hepatocellular carcinoma (HCC) cancer cell lines (Hep3B, HepG2, BEL-7404, SMMC-7721, Huh-7, PVTT-1, MHCC-97L, MHCC-97H and MHCC-LM3). (B) Typical immunohistochemical images of CCL2 staining in clinical HCC tissues and their matched normal tissues. Higher magnification images are shown under each panel (scale bars, 200 μm). Quantification of CCL2 protein staining in tumour and paraneoplastic tissues from patients with HCC in Cohort I (C) and Cohort II (D). The overall survival probabilities were calculated by the Kaplan-Meier method and analysed by the log-rank test for Cohort I patients (E) with low (n=31), moderate (n=39) or high (n=18) expression of CCL2 and Cohort II patients (F) with low/moderate (n=32) or high (n=27) expression of CCL2.
Moreover, the association between CCL2 expression in tumours and the postsurgery overall survival was investigated for 88/88 (100%) cases in Cohort I and 59/96 (61.5%) cases in Cohort II through univariate analyses. Lost cases were those with loss of follow-up information. For both cohorts, there was an inverse association between CCL2 expression and overall survival (figure 1E, F; log-rank p<0.0001 and p=0.0035, respectively). We also assessed whether CCL2 expression could serve as an independent predictor factor of patients’ overall survival in HCC Cohort I. We examined multiple clinicopathological variables, and found that tumour size and tumor-node-metastasis (TNM) stage, besides CCL2 expression, were associated with survival by univariate analysis (see online supplementary table S2). Next, a multivariate Cox proportional hazards analysis was performed, and those three positive predictor factors were adopted as covariates. We found tumour-derived CCL2 expression was an independent prognosticator of overall survival for HCC (CCL2 high, HR=7.199, p=0.0001; CCL2 modest, HR=3.092, p=0.029, see online supplementary table S2).
The growth of CCL2-overexpressing liver tumour cells is dependent on host CCR2
To establish the function of crosstalk mediated by CCL2/CCR2 signalling between tumour cells and the host microenvironment on liver cancer progression, the growth of three murine hepatoma cells, Hepa1-6, LPC-H12 and H22, in immunocompetent mice with wild type CCR2 or CCR2 knockout was evaluated. Hepa1-6, a cell line derived from BW7756 mouse spontaneous hepatoma that arose in a C57/L mouse, has pathological features similar to human HCC.26 LPC-H12 cells are a monoclonal line from a p53−/− and H-Ras knock-in HCC mouse tumour.27 The expression of CCR2 and secretion of its ligand, CCL2, was examined in these murine HCC cell lines. High levels of CCL2 were excreted in Hepa1-6 and LPC-H12 cells (figure 2A and see online supplementary figure S2A), but there was barely detectable expression of CCR2 on the cell membranes (figure 2B). By contrast, in H22 murine HCC cells, there was essentially no excretion of CCL2 and a modest expression of CCR2 (figure 2A, B). Growth of Hepa1-6, LPC-H12 and H22 cells was evaluated in orthotopic and subcutaneous murine HCC models with CCR2+/+ and CCR2−/− C57BL/6 mice (figure 2C). Germ line deletion of host CCR2 suppressed the orthotopic growth (figure 2D and see online supplementary figure S2C) and lung metastasis (see online supplementary figure S2B) of CCL2-overexpressing Hepa-6 and LPC-H12 cells, but had no effect on that of CCL2-negative H22 cells (figure 2E). Similarly, the tumour growth in CCR2−/− mice bearing Hepa1-6 subcutaneous tumours was reduced, relative to that in CCR2+/+ mice (figure 2F); this was not observed for H22 subcutaneous tumours (data not shown). These results indicate that CCL2/CCR2 signalling is involved in the growth of murine liver tumours overexpressing CCL2 and that blockade of CCL2/CCR2 signalling provides a therapeutic effect.

The growth of CCL2-overexpressing liver tumour cells is dependent on host CCR2. (A) Levels of chemokine CCL2 secreted by murine hepatocellular carcinoma (HCC) cell lines Hepa1-6, LPC-H12 and H22 were quantified in the supernatants using ELISA. Data are means±SEM. (B) Flow cytometric analysis of CCR2 expression in murine HCC cells, and human monocyte cells THP-1. (C) mRNA levels of CCR2 in the bone marrow (BM) and spleen (SP) of CCR2 wild type (WT) and knockout (KO) C57BL/6 mice. 1# and 2# indicate two different mice in each group. (D) Representative image of the Hepa1-6 orthotopic HCC tumours. At 3 weeks after implantation, tumour-bearing CCR2 KO mice exhibited smaller orthotopic liver tumours relative to CCR2 WT mice. The black dotted lines indicate the tumour regions (n=6 to 8 mice/group). (E) Representative image of the H22 orthotopic HCC tumours from CCR2 WT and KO mice (n=6 mice/group). (F) The subcutaneous tumour growth curve of Hepa1-6 in CCR2 WT and KO mice (n=8 mice/group).
Blocking CCL2/CCR2 signalling with a CCR2 antagonist suppresses liver tumour growth and postsurgical recurrence
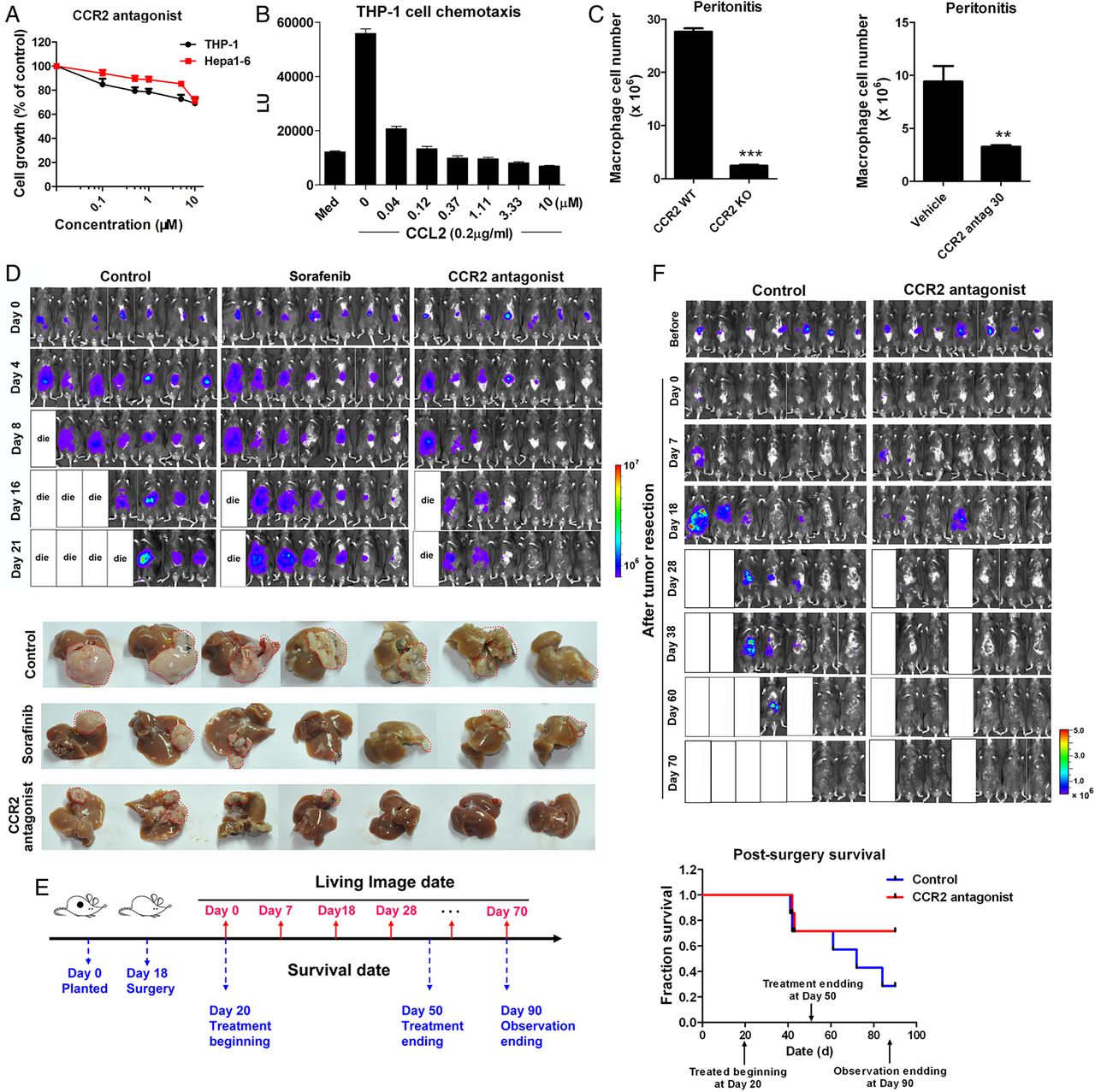
Due to the contribution of CCL2/CCR2 to the pathology of various inflammation-driven diseases, there have been attempts to develop CCR2 antagonists, anticipating therapeutic benefits from CCR2 inhibition.28 Here, the feasibility of targeting CCR2 for liver cancer therapy was evaluated with a CCR2 antagonist, RDC018, provided by GlaxoSmithKline. US patent US 8431590 B2 was issued for this compound class. This antagonist showed little cytotoxicity to Hepa1-6, LPC-H12 and H22 murine cancer cells or to human monocyte THP-1 cells, with IC50 values >10 μM (figure 3A and see online supplementary figure S3A). The antagonist inhibited CCL2-induced chemotaxis of THP-1 cells (EC50=2.89 nM; figure 3B). The present and other studies with CCR2 knockout mice demonstrated reductions of macrophage influx in the thioglycollate-induced peritonitis model (figure 3C, p<0.001). With this model, the antagonist caused a reduction in the number of infiltrated macrophages (65.4% inhibition at 30 mg/kg, p=0.005) in CCR2+/+ mice (figure 3C).
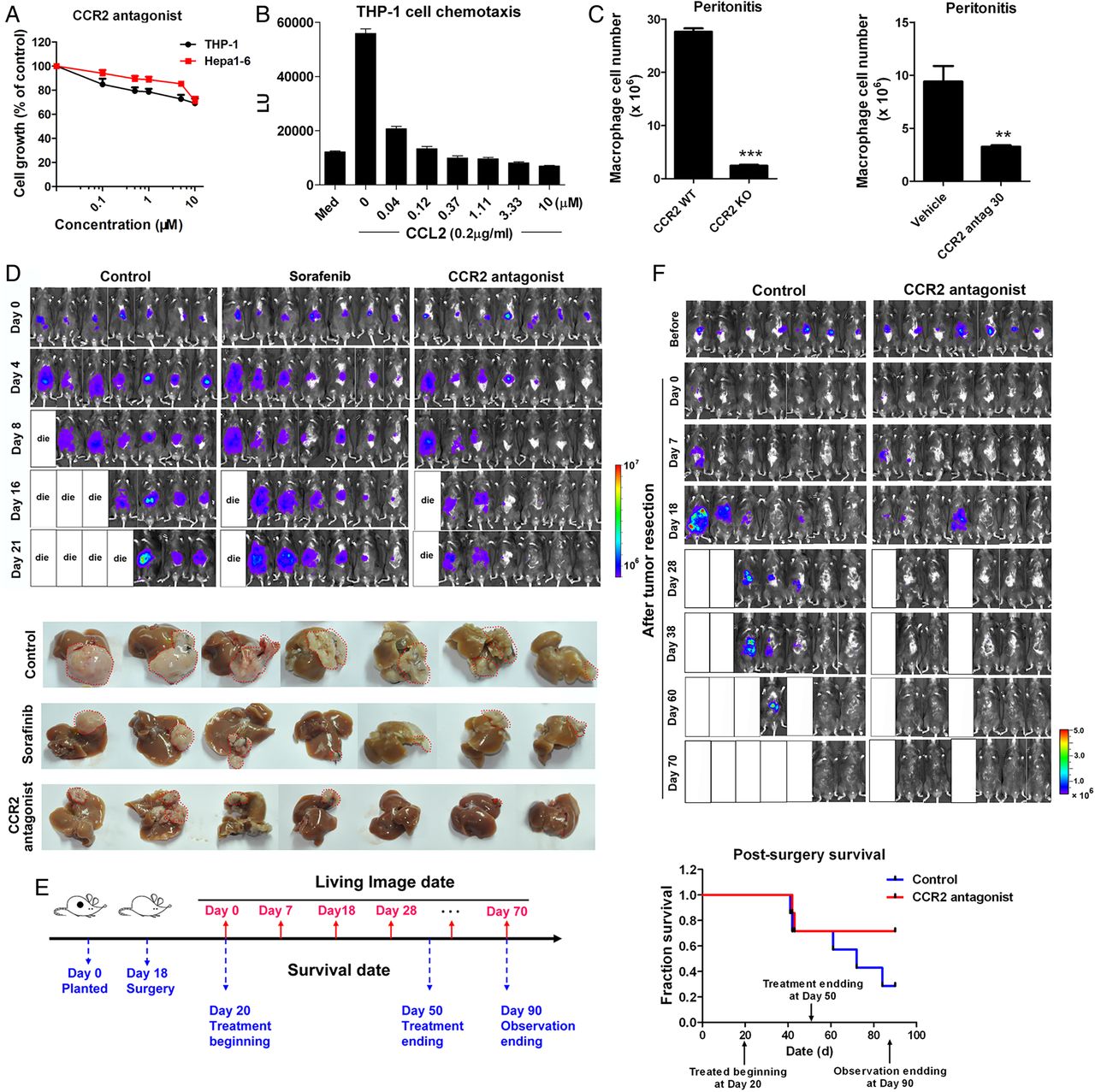
The CCR2 antagonist suppresses liver cancer growth and recurrence after tumour resection and prolongs survival of mice. (A) Growth of murine liver cancer cells Hepa1-6 and human monocytes THP-1after a 48 h of exposure to the CCR2 antagonist. (B) Inhibitory effect of the CCR2 antagonist on CCL2-mediated chemotaxis of THP-1 cells. (C) Quantification of intraperitoneal-infiltrated macrophages in the thioglycollate-induced murine peritonitis model in CCR2 wild type (WT)/knockout (KO) mice (left panel) or in mice treated with CCR2 antagonist (right panel). **p<0.01, ***p<0.001. (D) Representative bioluminescence images of mice treated orally with the CCR2 antagonist (30 mg/kg), sorafenib (30 mg/kg) or the vehicle, at the indicated times (n=7 mice/group). (E) The schedule of postsurgical liver cancer treatment and imaging. (F) Representative bioluminescence images and survival curves of mice (n=7) treated orally with the CCR2 antagonist (30 mg/kg) or vehicle.
The antitumour effect of blocking CCL2/CCR2 signalling with the CCR2 antagonist was assessed with an orthotopic liver tumour model (figure 3D). In mice receiving 30 mg/kg of the antagonist, tumour growth was inhibited. Further, the liver tumour volumes in treated mice were smaller than those in mice treated with vehicle or with sorafenib (30 mg/kg), a drug used clinically for liver cancer therapy (figure 3D and see online supplementary figure S3B). Moreover, the antagonist inhibited the abdominal dissemination and metastasis of cancer cells (see online supplementary figure S3C, D) and prolonged the survival of tumour-bearing mice, with no obvious effect on body weights or behaviours (see online supplementary figure S3E, F).
Although surgery is often chosen for treatment of HCC, recurrence frequently occurs and reduces the overall survival of patients.29 To explore approaches to preventing the recurrence of HCC after surgery, a surgical orthotopic HCC resection model was established (figure 3F). After surgery, the mice were randomised into control and CCR2 antagonist-treated cohorts. In the vehicle group, recurrent tumours and their peritoneal dissemination were evident at 7 days after resection (figure 3F). At 4 weeks, 71.4% of the control mice had intrahepatic relapse, relative to 28.6% in the treated group (figure 3F). Further, the overall survival rate of mice was improved after treatment (figure 3F).
The therapeutic effect of the CCR2 antagonist was further investigated with C57BL/6 mice bearing subcutaneous tumours expressing various levels of CCL2. Treatment with the CCR2 antagonist reduced the growth of CCL2-overexpressing Hepa1-6 tumours and LPC-H12 tumours (figure 4A and see online supplementary figure S4A, B), with inhibitions of 71.4% and 81.8%, respectively. A dose-dependent efficacy of the CCR2 antagonist (eg, 3 mg/kg, 10 mg/kg and 30 mg/kg) in a Hepa1-6 subcutaneous model also showed that 30 mg/kg of the antagonist provided the best therapeutic efficacy (see online supplementary figure S4C). However, there was no therapeutic effect of the CCR2 antagonist on H22 HCC tumours, which had essentially no expression of CCL2 (figure 4A and see online supplementary figure S4D), indicating that the therapeutic effect of the CCR2 antagonist was dependent on the excretion of tumour-derived CCL2. Moreover, based on observations of body weight, the CCR2 antagonist caused no observable toxic effects or loss of weight in these HCC mouse models (see online supplementary figure S4E–H).

CCL2/CCR2 blockade inhibits the growth of CCL2-overexpressing tumours, leading to reduced levels of blood Ly6C+ inflammatory monocytes and recruitment of tumour-associated macrophages (TAMs) and to enhanced peripheral and tumour-infiltrated CD8 lymphocytes. (A) Three murine hepatocellular carcinoma cells were separately transplanted into the right sides of C57BL/6 mice, which were orally dosed daily with the CCR2 antagonist (30 mg/kg) for the indicated number of weeks. Tumour volumes were measured (means±SEM; Hepa1-6, n=7; LPC-H12, n=5; H22, n=6; **p<0.01). (B) Representative images of subcutaneous tumours with vehicle or CCR2 antagonist treatment. (C) After CCR2-targeted therapy, the tumour-infiltrating immune cells and inflammatory cells, including B lymphocytes (CD19+), T lymphocytes (CD4+ or CD8+), T regulatory cells (CD4+CD25+Foxp3+), natural killer (NK) cells (NK1.1+) and TAMs (CD11b+Ly6G−F4/80+), were detected by flow cytometry in Hepa1-6 tumour tissues. *p<0.05, **p<0.01 relative to the vehicle group, (left panel). Representative flow cytometry data showing the proportion of CD4 and CD8 TILs and TAMs in Hepa1-6 tumour tissues of C57BL/6 mice (right panel). (D) H&E staining and immunohistochemistry (F4/80 and CD8) were performed on Hepa1-6 and LPC-H12 subcutaneous tumours after CCR2 antagonist treatment. Scale bar, 200 μm. (E) Flow cytometric analysis of blood leucocytes from C57BL/6 mice bearing Hepa1-6 tumours after treatment with the CCR2 antagonist. Percentage of peripheral T lymphocytes (CD4+ or CD8+) and monocytes (Ly6Chigh, CD11b+Ly6ChighF4/80−CCR2+; Ly6Clow, CD11b+Ly6ClowF4/80−CCR2+) are shown (*p<0.05, **p<0.01, left panel). Representative flow cytometric images of blood CD4 and CD8 lymphocytes and Ly6Chigh and Ly6Clow monocytes (gated as CD11b+ Ly6C+ cells) in control and treated tumour-bearing mice (right panel). TIL, tumour-infiltrating lymphocyte.
Treatment with the CCR2 antagonist leads to reduced blood Ly6C+ inflammatory monocytes, recruitment of TAMs, and enhanced peripheral and tumour-infiltrated CD8 lymphocytes in a tumour-derived CCL2-dependent manner
For mouse and human tumours, CCL2 is the major tumour-derived factor responsible for recruiting monocytes/macrophages, and the CCL2/CCR2 axis connects the crosstalk between tumour cells and the associated stroma.30 ,31 To investigate the antitumour mechanism of the CCR2 antagonist, the numbers of tumour-infiltrating immune and inflammatory cells were measured in Hepa1-6, LPC-H12 and H22 subcutaneous tumours. Flow cytometric analysis of Hepa1-6 tumours showed a 71.4% reduction of TAMs and a 2.52-fold increase of CD8+ T cells in mice treated with the CCR2 antagonist (figure 4C). The numbers of other inflammatory cells, such as regulatory T cells, B cells and natural killer (NK) cells, exhibited no appreciable changes, except for a slight increase of CD4+ T cells (figure 4C). Immunohistochemical evaluations also showed a decrease of F4/80+ TAMs and an increase of CD8+ T cells in Hepa1-6 and LPC-H12 tumours (figure 4D) after treatment with the CCR2 antagonist but not in H22 tumours (see online supplementary figure S5A). Similarly, lower numbers of infiltrated TAMs and increased numbers of cytotoxic CD8+ and CD4+ T cells were also observed in the Hepa1-6 subcutaneous tumours of CCR2−/− mice, relative to that in CCR2+/+ mice (see online supplementary figure S5B). The effects of the CCR2 antagonist on mononuclear phagocytes in blood and spleen of these tumour-bearing mice were determined. Treatment with the CCR2 antagonist caused a decrease in the number of blood Ly6Chigh inflammatory monocytes and a slight decrease of Ly6Clow non-inflammatory monocytes, but increased the numbers of CD8+ and CD4+ T cells in Hepa1-6 (figure 4E) and LPC-H12 HCC models (see online supplementary figure S5C). On the contrary, the percentages of blood CD4+ and CD8+ T cells were not affected, but there was an increase in the number of inflammatory monocytes in the treated mice bearing H22 tumours (see online supplementary figure S5D). No appreciable changes of these cell subsets were found in the spleens of the three models (see online supplementary figure S5E–G).
Increased cytotoxic CD8 T lymphocytes are responsible for the antitumour activity of CCR2 antagonist
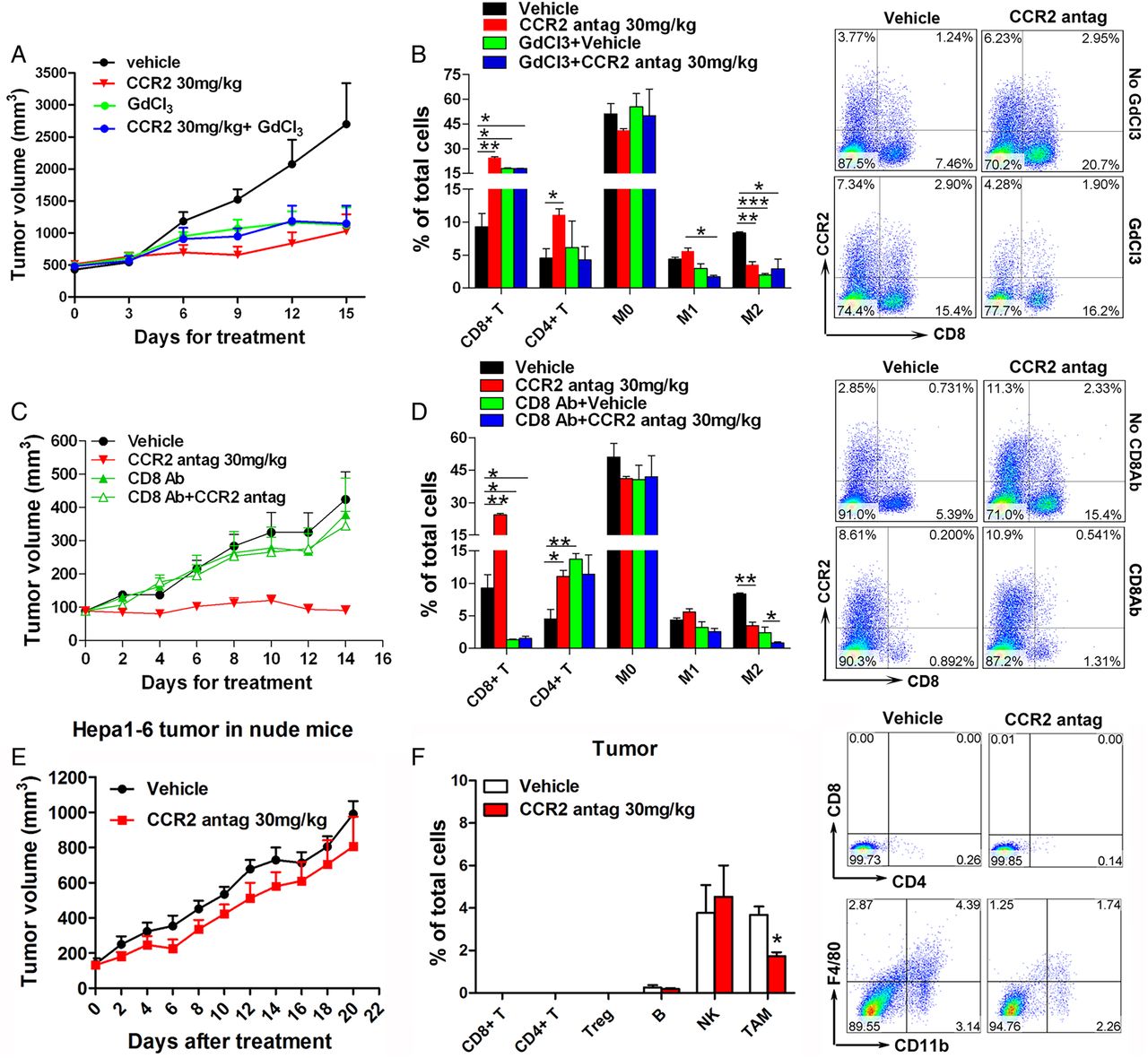
The results discussed above raised the question whether TAMs and CD8+ tumour-infiltrating lymphocytes (TILs) are involved in the antitumour activity of the CCR2 antagonist. To elucidate the role of macrophage depletion in the antitumour activity of the CCR2 antagonist, its effects were compared with the monocyte/macrophage depleting agent, GdCl3.32 ,33 C57BL/6 mice were inoculated with Hepa1-6 cells to form subcutaneous tumours and subsequently treated with GdCl3, with the CCR2 antagonist, or with the combination. Tumour growth in GdCl3-treated mice was inhibited, similar to that in CCR2 antagonist-treated group (figure 5A). Treatment with the combination did not result in an additive or synergistic antitumour effect (figure 5A). The proportions of inflammatory cells in the tumour tissues of each group were assessed. Macrophage depletion with GdCl3 inhibited the infiltration of M1-TAMs (CD11b+/F4/80+/CD206−) and M2-TAMs (CD11b+/F4/80+/CD206+), but treatment with the CCR2 antagonist suppressed the number of M2-TAMs (figure 5B). Treatment with the CCR2 antagonist, GdCl3, or the combination increased the proportion of cytotoxic CD8+ TILs in the tumour microenvironment (figure 5B), but there were no substantial changes in other inflammatory cells (B lymphocytes, NK cells or myeloid-derived suppressor cells; see online supplementary figure S6A). These results indicated that the CCR2 antagonist acts through blocking M2-TAM infiltration and activating the CD8+ T cell immune system.

Depletion of CD8+ T cells or T cell immunodeficiency abolishes the antitumour effect of the CCR2 antagonist. (A) C57BL/6 mice (n=7) were inoculated subcutaneously with Hepa1-6 tumours and subsequently treated with GdCl3 (to deplete macrophages) with or without the CCR2 antagonist. Tumour growth was measured. (B) The relative percentages of the tumour-infiltrating inflammatory cells, as determined by fluorescence-activated cell sorting (FACS), in the tumour tissues of mice subjected to various treatments (left panel). These include CD4+ or CD8+ TILs; M0-TAM, CD11b+Ly6G−F4/80−; M1-TAM, CD11b+Ly6G−F4/80+CD206−; and M2-TAM, CD11b+Ly6G−F4/80+CD206+ cells. Representative flow cytometry gating images showing the proportion of CD8+ TILs in Hepa1-6 tumour tissues after treatment with either the CCR2 antagonist or GdCl3 or both (right panel). (C) C57BL/6 mice (n=7) bearing subcutaneous Hepa1-6 cells were treated with CD8-neutralising antibodies (to deplete CD8+ T cells) with or without the CCR2 antagonist. (D) After the mice received various treatments, their tumours were measured, and the ratios of TAM and CD4 and CD8 TILs were quantified by FACS (left panel). Contour plots showing the percentage of CD8+ TILs in subcutaneous tumours after treatments (right panel). (E) Volumes of Hepa1-6 tumours in nude mice after treatment with 30 mg/kg of the CCR2 antagonist at the indicated days (n=7). (F) Flow cytometric analysis of the tumour-infiltrating inflammatory cells in Hepa1-6 tumour-bearing nude mice after treatment with the CCR2 antagonist (left panel). Representative flow cytometry gating images showing the proportion of TAMs and T lymphocytes (right panel). Data are presented as means±SEM (*p<0.05, **p<0.01 and ***p<0.001). GdCl3, gadolinium chloride; TAM, tumour-associated macrophage; TIL, tumour-infiltrating lymphocyte.
To establish the role of cytotoxic T cells in this process, cytotoxic CD8+ T cells were depleted with CD8-neutralising antibody. As expected, the amounts of CD8+ T lymphocytes in the peripheral blood of mice treated with the CD8 antibody were lower than those in the untreated group (see online supplementary figure S6B, C). Depletion of CD8+ T cells diminished the antitumour effect of the CCR2 antagonist (figure 5C), and, correspondingly, few CD8+ TILs were found in the tumour tissues of mice treated with the vehicle or with the CCR2 antagonist (figure 5D). After blocking CD8+ T cells, there was still a decrease of infiltrated M2-TAMs in the tumour tissues of mice treated with the CCR2 antagonist (figure 5D), indicating that CD8+ cytotoxic T cells are a downstream target of TAMs in mice dosed with the CCR2 antagonist. There were no obvious changes in other tumour infiltrating inflammatory cells (B lymphocytes, NK cells or myeloid-derived suppressor cells) after treatment with the CCR2 antagonist alone or combined with CD8-neutralising antibody (see online supplementary figure S6D). To better confirm the involvement of T lymphocyte immune in the antitumor effect of CCR2 antagonist, a nude mice model bearing Hepa1-6 subcutaneous tumours was used. As expected, treatment with the CCR2 antagonist had no effect on the growth of Hepa1-6 tumours (figure 5E and see online supplementary figure S6E) or on body weights of nude mice (see online supplementary figure S6F). Flow cytometric analysis of the Hepa1-6 tumours showed that recruitment of TAMs was still inhibited after treatment with the CCR2 antagonist, although no CD4 or CD8 TILs were detected in the tumour tissues (figure 5F).
Blockade of CCL2/CCR2 abolishes the crosstalk between tumour cells and macrophages and suppresses M2 macrophage polarisation and its immunosuppression of lymphocytes
Macrophages have functional plasticity, with the capacity to change their profiles in response to different tumour-educated microenvironments.7 ,10 ,34 Relative to M1 macrophages, M2 macrophages produce higher amounts of interleukin (IL) 10, arginase-1, and CD206, but express lower levels of IL-12 and inducible inducible nitric oxide synthase (iNOS).7 ,10 In view of the selective inhibition of the CCR2 antagonist on the amount of M2-TAMs in the tumour microenvironment, we determined if blocking of the CCL2/CCR2 axis mediates the functional polarisation. The M1-TAM and M2-TAM cells in Hepa1-6 tumour tissues (figure 6A) were discriminated by use of classical markers of M1 and M2 macrophages and their respective secretion spectra. Quantitative PCR analyses of the sorted cell populations confirmed expression of the classical markers of M1 and M2 macrophages in M1-TAMs and M2-TAMs, respectively (figure 6B). Relative to M1-TAMs in tumour tissue, M2-TAMs produced more soluble factors (IL-6, IL-10, granulocyte-colony stimulating factor (G-CSF), keratinocyte-derived chemokine (KC), CCL2, macrophage inflammatory proteins-1 (MIP-1) and MIP-2; figure 6B). After treatment of mice with the CCR2 antagonist, most of the cytokines or chemokines produced by M2-TAMs were decreased in the TAMs (figure 6C). Similar decreases of M2-TAM-produced cytokines were found in the ascites of Hepa1-6 orthotopic liver cancers (see online supplementary figure S7A) and in subcutaneous tumours treated with the CCR2 antagonist (see online supplementary figure S7B).

Blockade of CCL2/CCR2 abolishes the crosstalk between tumour cells and macrophages and suppresses M2 macrophage polarisation and its immunosuppression of lymphocytes. (A) Flow cytometry gating strategies used to define M1-TAMs (CD11b+/F4/80+/CD206−) and M2-TAMs (CD11b+/F4/80+/CD206+). Macrophages were sorted from subcutaneous tumours of mice without CCR2 antagonist therapy. mRNA expression of classic and recognised markers for M1 and M2 macrophages was determined in these two subsets of sorted macrophages. (B) Cytokine and chemokine profiles of the indicated macrophages. (C) The CD11b+/F4/80+ macrophages were sorted from subcutaneous tumours of mice receiving vehicle or 30 mg/kg of the CCR2 antagonist. Cytokine and chemokine profiles of each subset of macrophages were determined. Data were presented as means±SEM for seven mice in each group. (D) Bone marrow derived macrophages (BMDMs) were exposed to conditioned medium (CM) of Hepa1-6 tumour cells and subsequently treated with vehicle, the CCR2 antagonist (CCR2 antag, 1 μM or 5 μM), CCL2 neutralising antibody (CCL2 Ab, 1 μg/mL) or isotype control antibody for 48 h. qRT-PCR results showing the expression of M1 and M2 macrophage-specific markers in BMDM with various treatments. (E) The mRNA expressions of indicated genes in BMDM were determined after treatment. (F) Effect of CCL2/CCR2 blockade on the splenic CD8+ T lymphocyte suppression caused by murine hepatocellular carcinoma (HCC) cell-educated BMDMs. Representative plots of carboxyfluorescein succinimidyl ester (CFSE) signal intensity of CD8+ T cells are depicted for inactivated CD8+ T or anti-CD3/CD28-activated CD8+ T cells followed by coculturing with normal BMDMs (M0), or Hepa1-6 tumour cell educated-BMDM (Mh) with or without CCL2/CCR2 blockade by CCR2 antagonist or CCL2-neutralising antibody (left panel). The quantification of proliferated CD8+ T cells in different groups (right panel). Data are presented as means±SEM from three independent experiments (*p<0.05, ***p<0.001). TAM, tumour-associated macrophage.
To determine if liver cancer cells and CCL2/CCR2 inhibition regulate M2 macrophage polarisation, we used a cell-based assay, in which murine BMDMs were exposed to the CM of Hepa1-6 cells to mimic the microenvironment interactions. After exposure of BMDMs to the CM, the expression of M2 marker genes, arginase-1 and IL-10, were increased, and expression of M1 genes, inducible NOS and IL-12, were decreased (figure 6D). Blocking CCL2/CCR2 signalling by the CCR2 antagonist or by CCL2-neutralising antibody reversed this effect (figure 6D). Moreover, the increased M2-type cytokines (IL-6, G-CSF, MIP-1 and MIP-2) induced by Hepa1-6 CM were reduced after CCR2 inhibition (figure 6E). These results indicate that blocking CCL2/CCR2 signalling destroys the crosstalk between tumour cells and TAMs, remodels the tumour-educated microenvironment, and inhibits the M2 polarisation of TAMs.
In a coculture system with BMDMs and splenocytes, tumour-educated BMDMs inhibited splenic CD8+ T cell proliferation after treatment with anti-CD3/CD28 antibody (figure 6F). CCL2/CCR2 blockade by the CCR2 antagonist or by CCL2-neutralising antibody alleviated the immunosuppressive activity of tumour-educated macrophages (figure 6F). Similarly, knockout of CCR2 in BMDMs attenuated the T cell immunosuppression resulting from the murine tumour-educated BMDMs (data not shown). Moreover, a modest reversion of CD4+ T cell suppression was observed after CCL2/CCR2 blockade (see online supplementary figure S8).
To understand the clinical implications of these interactions, we analysed the expression of CCL2, CD68 and CD8 in tumour tissues from 85/88 (96.6%) cases in Cohort I to determine the distribution of CCL2-expressed HCC cells, intratumorous CD68+ TAMs and CD8+ TILs. The expression of CCL2 in tumour cells correlated with the tumour-infiltrating TAM and CD8+ T cells (figure 7A, B). For patients with higher levels of CCL2 expression in their tumour tissues, the tumour-infiltrating CD68+ TAMs were increased, and the intratumorous CD8+ TILs were lower (figure 7A, B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In hepatocellular carcinomas (HCCs), high CCL2 expression correlates with more tumour-infiltrated tumour-associated macrophages and fewer CD8+ TILs. (A) The 85 surgical specimens of HCCs in Cohort I were immunostained with antibodies to CCL2, CD68 and CD8. Representative stains from the same tumour samples are shown. Scale bar, 200 μm. (B) The ratios of low or high level of CD8 TILs staining in tumours with low (n=27), moderate (n=34) or high (n=24) levels of CCL2 expression. (C) Schematic depiction of the reconstruction of the tumour microenvironment by CCL2/CCR2 blockade with the CCR2 antagonist. TIL, tumour-infiltrating lymphocyte.
Thus, in the HCC tumour microenvironment, tumour cells secrete CCL2 to recruit CCR2+ monocytes/macrophages and educate TAMs to promote its M2-type polarisation and immunosuppression. The educated TAMs enhance the production of various immunosuppressive cytokines and chemokines (IL-6, KC, G-CSF, MIP-1 and MIP-2) and suppress the proliferation of cytotoxic CD8+ TILs, directly or indirectly. Blocking the CCL2/CCR2 axis with a CCR2 antagonist reduces TAM recruitment and M2 phenotype polarisation and thus alleviates the CD8+ T cell immunosuppression and enhances the tumour immunotherapeutic effect (figure 7C).
Discussion
Chemokines and their receptors are highly interesting therapeutic targets for pharmaceutical and biotechnology companies. Here we show that CCL2 is overexpressed in human HCCs and is associated with a poor prognosis for patients. Blockade of CCL2/CCR2 with a CCR2 antagonist inhibits tumour growth and metastasis, prevents postsurgical recurrence, and, in syngeneic murine HCC models, enhances survival in a tumour-derived CCL2-dependent manner. CCR2 blockade reduces blood inflammatory monocytes and infiltration of intratumorous TAMs, especially the M2-type TAMs, and increases the number of peripheral CD8+ T cells and cytotoxic CD8+ TILs in the murine microenvironment. Depletion of CD8+ T cells or T cell immunodeficiency abolishes the antitumour activity of the CCR2 antagonist. Moreover, in various mouse models, blocking of CCR2 with the CCR2 antagonist reduces the number of M2-type TAMs and the production of M2-type cytokines (IL-6, CCL2, KC, G-CSF, MIP-1 and MIP-2). This phenomenon is modelled in a coculture system in which there was induction of M2 polarisation and enhanced immunosuppression by murine tumour-secreted factors and reversal by the CCR2 antagonist.
There are clinical implications of these findings. First, CCL2 is overexpressed and associated with poor survival of patients with HCC and the CCL2/CCR2 axis is necessary for liver cancer growth and metastasis. These results support the concept that the CCL2/CCR2 axis is a new therapeutic target for patients with HCC overexpressing CCL2. Recently, Chew et al found that CCL2 is secreted in tumour-infiltrated immune cells such as NK cells but not tumour cells, and higher TIL-derived CCL2 mRNA levels were correlated with patients with longer HCC survival via univariate analysis.35 The opposing results might result from different CCL2-secreting donor cells and their varied function in the tumour microenvironment. Second, the CCR2 antagonist targets inflammatory monocytes/TAMs, inhibits macrophage M2-polarisation and overcomes the immunosuppressive effect of TAMs on cytotoxic T lymphocytes. Unlike those in tumour cells, the genomes of macrophages are stable, suggesting that, in immunotherapeutic approaches, TAMs may not readily become drug resistant.5 ,36 Since HCC is an aggressive cancer with limited effective treatment options,1 there is a need for new therapies, and immunotherapy is being investigated as a strategy.37 ,38 Of note, a phase I/II trial showed a promising antitumour response of nivolumab, a clinically used programmed cell death protein-1 (PD-1). Inhibitor, in patients with advanced HCC.39 A requirement for immunotherapeutic approaches against cancer is the expansion and entry of tumour antigen-specific T cells into the tumour microenvironment and the removal of the inhibitory pathways such as PD-1/PD-L1 axis that limit T cells activation and responses.40–42 However, established tumours have developed an anti-inflammatory stroma and recruit immunosuppressive cells to the tumour sites, both of which prevent immune eradication, even when tumour antigen-specific T cells are in the periphery.40 As immunosuppressive cells in the liver tumour microenvironment,36 TAMs are increased and of prognostic significance in patients with HCC after surgical resection.9 ,43 ,44 There are four main strategies for TAM-based antitumour therapy: inhibiting macrophage recruitment, suppression of TAMs survival, enhancing the M1 tumoricidal activity of TAMs and blocking the M2 tumour-promoting activity of TAMs.36 ,45 Here, our results show that targeting CCL2/CCR2 signalling with the CCR2 antagonist reduces monocyte/TAM recruitment and M2 phenotype polarisation and thus alleviates CD8+ T cell immunosuppression and enhances tumour immunotherapeutic effect. For inflammatory diseases, CCL2/CCR2 inhibitors such as the CCR2 antagonist and CCL2 neutralising antibodies have been used to inhibit monocyte/macrophage recruitment.22 ,28 ,45 Recently, Bonapace et al46 reported that cessation of CCL2 neutralised antibody accelerates breast cancer metastasis by promoting angiogenesis and reinfiltration of monocytes. In our postsurgical recurrence models, after withdrawal of the CCR2 antagonist treatment, there is no obvious malignant progression observed, and animals receiving CCR2 inhibition have a much longer survival rate, suggesting the different outcomes between the CCL2 or CCR2 targeting strategy. Since genotoxic chemical exposure, non-alcoholic fatty liver and chronic HBV/HCV infection are major contributors to increased HCC incidence,47 it is interesting to figure out the role of the CCL2/CCR2 axis and its therapeutic value in liver fibrosis and hepatocarcinogenesis with relative preclinical murine models in future studies.
In conclusion, we have uncovered a new therapeutic strategy for targeting tumour-infiltrating macrophages in HCCs via CCL2/CCR2 signalling. Blocking the CCL2/CCR2 axis with the CCR2 antagonist reduces monocytes/TAMs recruitment and M2 phenotype polarisation and thus alleviates the CD8+ T cell immunosuppression and enhances the tumour immunotherapeutic effect. A multifaceted approach that employs CCL2/CCR2 targeted immunotherapy in combination with other established HCC therapies should prove more effective in alleviating this malignant disease.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online tables
Footnotes
Correction notice This article has been corrected since it published Online First. The funding statement has been updated.
Acknowledgements The authors thank Dr Donald L Hill for editorial assistance.
Contributors XL and HW conceived and designed experiments. XL and HW analysed data and wrote the manuscript. HW supervised the project. XL, WY, YY, PC and JL performed the in vitro and in vivo studies. Pathology support, tissue provision and patients' information collection were accomplished by BL and XJ. RC, HS and DX assisted in experimental design and data evaluation. All authors reviewed and approved the manuscript.
Funding This work was supported by grants from the Ministry of Science and Technology of China (2014AA020524), the National Natural Science Foundation of China (91529305, 81125020, 81427805, 81302809 and 81502122), the Instrument Developing Project of the Chinese Academy of Sciences, the Science and Technology Commission of Shanghai Municipality (12431900500 and 14391901800), the China Postdoctoral Science Foundation (2014M551469), the CAS-SIBS Postdoctoral Foundation (2014KIP315), and the Key Laboratory of Food Safety Research of INS, SIBS, CAS.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Internal Review and Ethics Boards of the Shanghai Eastern Hepatobiliary Surgery Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.