Article Text

Abstract
Objective The gut microbiota has been implicated as an environmental factor that modulates obesity, and recent evidence suggests that microbiota-mediated changes in bile acid profiles and signalling through the bile acid nuclear receptor farnesoid X receptor (FXR) contribute to impaired host metabolism. Here we investigated if the gut microbiota modulates obesity and associated phenotypes through FXR.
Design We fed germ-free (GF) and conventionally raised (CONV-R) wild-type and Fxr−/− mice a high-fat diet (HFD) for 10 weeks. We monitored weight gain and glucose metabolism and analysed the gut microbiota and bile acid composition, beta-cell mass, accumulation of macrophages in adipose tissue, liver steatosis, and expression of target genes in adipose tissue and liver. We also transferred the microbiota of wild-type and Fxr-deficient mice to GF wild-type mice.
Results The gut microbiota promoted weight gain and hepatic steatosis in an FXR-dependent manner, and the bile acid profiles and composition of faecal microbiota differed between Fxr−/− and wild-type mice. The obese phenotype in colonised wild-type mice was associated with increased beta-cell mass, increased adipose inflammation, increased steatosis and expression of genes involved in lipid uptake. By transferring the caecal microbiota from HFD-fed Fxr−/− and wild-type mice into GF mice, we showed that the obesity phenotype was transferable.
Conclusions Our results indicate that the gut microbiota promotes diet-induced obesity and associated phenotypes through FXR, and that FXR may contribute to increased adiposity by altering the microbiota composition.
- BILE ACID
- OBESITY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The gut microbiota contributes to obesity
The gut microbiota modulates farnesoid X receptor (FXR) signalling
The gut microbiota metabolises a naturally occurring FXR antagonist
What are the new findings?
The gut microbiota promotes obesity and associated diseases through FXR
FXR alters the gut microbiota
The altered gut microbiota contributes to the obese phenotype
How might it impact on clinical practice in the foreseeable future?
Understanding how the gut microbiota modulates bile acids and signalling through FXR to modulate host metabolism may provide novel insights into how the microbiota can be targeted to prevent obesity.
Introduction
The gut microbiota is considered an environmental factor that modulates host metabolism, and recent evidence suggests that it contributes to the development of obesity and metabolic diseases. For example, the gut microbiota is altered in obese mice and humans1–5 as well as in humans with type 2 diabetes.6 ,7 Furthermore, germ-free (GF) mice have reduced adiposity and obesity, and transfer of microbiota from obese conventionally raised (CONV-R) mice to GF recipients has revealed that obesity is a transmissible trait through the microbiota.8–11 Recent data also showed that faecal microbiota transplants from lean glucose-sensitive human donors improve insulin sensitivity in humans with the metabolic syndrome,12 indicating that an altered gut microbiota may directly contribute to metabolic disease. However, little is known about the underlying mechanisms by which the gut microbiota affects host metabolism.
The gut microbiota metabolises dietary and host-derived molecules to produce bioactive components, which may play a role in host metabolism.13 Bile acids, for example, are synthesised in the liver from cholesterol where they are conjugated to glycine (human) or taurine (mouse), and metabolised into secondary bile acids in the gut by the microbiota.14 ,15 Bile acids are now recognised as signalling molecules that act through the nuclear receptor farnesoid X receptor (FXR) and the G-protein-coupled receptor TGR5 to regulate lipoprotein and glucose metabolism.16–21 Fxr-deficient mice are protected against diet-induced obesity,22 and studies in tissue-specific Fxr-deficient mice have revealed that intestinal FXR is required for the development of obesity, insulin resistance and non-alcoholic fatty liver disease in response to a high-fat diet (HFD).23 ,24
By comparing GF and CONV-R mice, we recently identified tauro-beta muricholic acid (TβMCA) as a potent FXR antagonist and showed that FXR suppression is alleviated by microbial metabolism of TβMCA to βMCA.15 Similarly, modulation of the mouse gut microbiota using antibiotics or the antioxidant tempol increases the levels of TβMCA and suppresses FXR activity.15 ,23 ,24 Furthermore, a recent study in human beings has shown that vancomycin treatment results in reduced Gram-positive bacteria, altered bile acid metabolism, and decreased insulin sensitivity.25 These studies suggest that changes in the bile acid composition, and thus altered FXR signalling, may be a link between alterations in the microbiota and impaired host metabolism.
Here we use GF and Fxr−/− mice to test the hypothesis that the gut microbiota promotes obesity and associated phenotypes by activating FXR signalling. In addition, we investigate if FXR signalling contributes to obesity-associated phenotypes by altering the gut microbiota.
Methods
Mice and diet
Male wild-type and whole-body Fxr-deficient GF and CONV-R mice on C57Bl/6 genetic background were maintained initially on autoclaved chow diet (LabDiet, Diet-5021-3-BG) and had access to diet and water ad libitum. GF mice were housed according to standard protocols in sterile flexible film isolators.26 At 9–14 weeks of age, the mice were switched to irradiated vacuum-packed HFD (Harlan Laboratories, Diet TD.96132; 40% of energy from fat) ad libitum and then weighed in the isolators weekly for 10 weeks. Mice were housed under a 12 h light–dark cycle at 20±2°C and humidity range of 50±15%. All procedures were approved by the Ethics Committee on Animal Care and Use in Gothenburg, Sweden.
Glucose and insulin tolerance tests
An oral glucose tolerance test was performed after 10 weeks on HFD. Following a 4 h fast, mice were removed from the isolators and orally gavaged with 20% d-glucose (3 g/kg body weight). Blood was drawn from the tail vein at −30, 0, 15, 30, 60, 90 and 120 min and blood glucose levels were measured using a HemoCue glucometer. In addition, blood was collected from the tail vein at 0, 15 and 30 min for analysis of serum insulin levels using Ultra Sensitive Mouse Insulin ELISA (Crystal Chem).
An insulin tolerance test was performed in an independent cohort of mice after 10 weeks on HFD. Following a 4 h fast, mice were removed from the isolators and injected intraperitoneally with 0.25 U insulin/mL (0.75 mU/g body weight). Blood was drawn from the tail vein at −30, 0, 15, 30, 60, 90 and 120 min, and blood glucose levels were measured using a HemoCue glucometer.
Immunohistochemistry of pancreas and adipose tissue
Antibodies used are listed in online supplementary table S1. Paraffin-embedded pancreatic tissues fixed in 4% paraformaldehyde (HistoLab Products) were sectioned (6 μm). For the analysis of beta-cells, sections were stained with a guinea pig anti-insulin antibody and Vectastain ABC reagent followed by colour development using Vulcan Fast Red according to the manufacturer's protocol and counterstained with haematoxylin. Islet size and beta-cell area were quantified in two sections/mouse at least 200 µm apart from 5–7 mice/group using the software BioPix IQ Classic. Beta-cell mass was calculated by multiplying beta-cell area by the weight of the pancreas.
Supplementary materials
Paraffin-embedded epididymal white adipose tissue (WAT) was sectioned (6 μm) (HistoLab Products) and stained for MAC2 and crown-like structures as described previously.27 The crown-like structures were manually counted with the software MIRAX Viewer in 14–20 images/histological section per mouse.
Quantitative real-time PCR
Approximately 30 mg epididymal WAT and liver were homogenised with TissueLyzer II (Qiagen) followed by total RNA extraction with RNeasy Mini Kit (Qiagen). Five hundred nanograms of pure cDNA were synthesised using the High Capacity cDNA Reverse Transcription Kit according to the manufacturer's protocol (Applied Biosystems). The cDNA was diluted 7× before subsequent reactions. For qRT-PCR reactions, IQ SYBR Green Supermix (Bio-Rad) was used at an end volume of 25 µL, and 0.9 µM of gene-specific primers were added (see online supplementary table S2). Gene-specific primers were normalised to the housekeeping gene L32. Each primer set was verified by analysis of their melt curves, and the assays were performed in a 7900HT Fast Real-Time PCR System (Applied Biosystems) or CFX96 Real System (Bio-Rad). The ΔΔCT analysis method was used to analyse the reactions.
Supplementary materials
Liver lipid analysis
Snap frozen tissue was placed into 2 mL Sarstedt tubes loaded with six zirconium oxide beads (3 mm) (Retsch). Tissue was homogenised for 10 min at 25 Hz in 500 µL methanol using a Mixer Mill 301 instrument (Retsch). Lipids were then extracted using the Folch method.28 Cholesteryl esters and triglycerides were quantified using direct infusion/mass spectrometry as described previously.29
Alanine transaminase analysis
Alanine transaminase (ALT) activity was measured in 20 µL vena cava sera from n = 5–8 mice/group using EnzyChrom ALT Assay Kit (Bioassay Systems). The samples were added to CFX96 Real System plates (Bio-Rad), and the ALT activity was thereafter determined according to colorimetric measurement using OD read at 340 nm.
Microbiota analysis
Total DNA was isolated from faecal samples after 10 weeks of HFD feeding as described previously.30–32 The V4 region of the 16S rDNA gene was amplified using barcoded primers, and sequencing was performed using MiSeq Gene & Small Genome Sequencer from Illumina. Reads were analysed using the MacQIIME V.1.8 software package (http://www.wernerlab.org/software/macqiime33) as described previously.34 Briefly, operational taxonomic units were picked using UCLUST, and taxonomic assignments were made using the Ribosomal Database Project. Singletons, chimeric sequences and those that failed to align with PyNAST were removed. Relative abundance, Unifrac α and beta diversity were calculated and phylogeny constructed using UPGMA (Unweighted Pair Group Method with Arithmetic Mean). Significance of differences in abundances was calculated using t test and false discovery-rate correction. Linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed using standard parameters (p<0.05 and LDA score 2.0) as described in ref. 35.
Bile acid analysis
Bile acids were extracted from vena cava serum (n=5–9 mice/group) and from caecal content (n=3–9 mice/group) after 10 weeks of HFD using 10 volumes of methanol-containing internal standards. After vigorous mixing for 5 min, the samples were centrifuged at 22 000 g for 10 min. The supernatant was removed and diluted 10 times with water/methanol (1:1). The samples were then analysed using ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS).36
Microbiota transfer experiments
Caecal content was collected from wild-type and Fxr−/− mice on HFD for 10 weeks; at the time of collection, mice were fed and the light had been on for 3–5 h. GF C57BL/6J mice aged 9 weeks were gavaged with the caecal content and transferred to HFD for 10 weeks. Mice were weighed weekly. After 10 weeks of HFD, body fat was assessed by MRI and an oral glucose tolerance test was performed in these mice.
Statistical analysis
Data are represented as mean ±SEM. Multiple groups were analysed by two-way ANOVA followed by Bonferroni post hoc test; detailed information can be found in online supplementary table S3. Statistical differences in bile acids between wild-type and Fxr−/− mice were determined using two-tailed, unpaired, FDR-corrected Student's t test. GraphPad Prism 5 software was used for two-way ANOVA and t test. The effect of Fxr knockout on weight gain over time was analysed by using a mixed two-way ANOVA test in R. In this test, the genotype was considered as a between-sample variable and time as a within-sample variable.
Supplementary materials
Results
Effect of the gut microbiota on diet-induced obesity and glucose metabolism is reduced in Fxr−/− mice
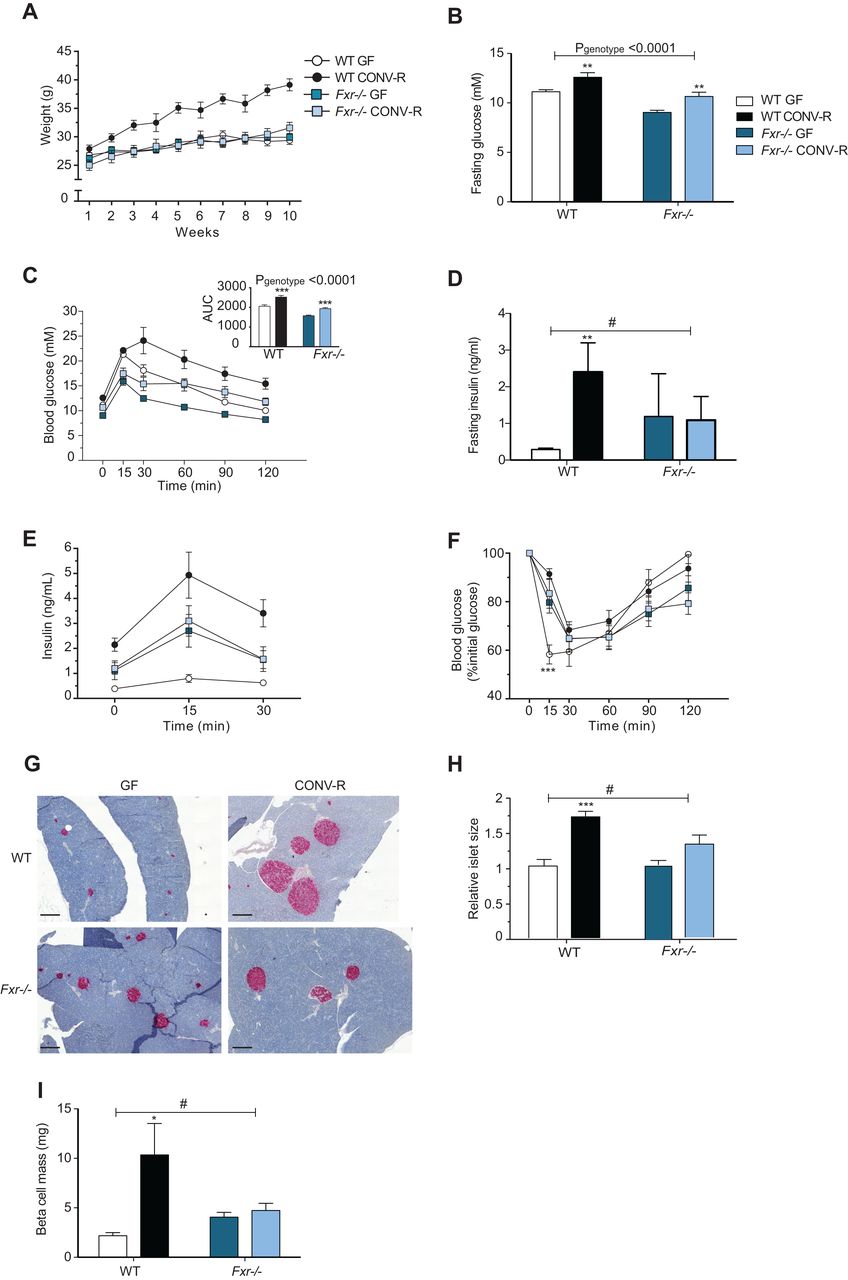
Previous studies have shown that GF mice are protected against diet-induced obesity and have improved glucose metabolism compared with their colonised counterparts,9 ,37 ,38 but the underlying mechanisms are unknown. Here, we fed GF and CONV-R wild-type and Fxr−/− mice a HFD for 10 weeks to investigate whether the gut microbiota affects diet-induced weight gain and glucose metabolism through FXR. As expected, CONV-R wild-type mice gained significantly more weight than GF wild-type mice after 10 weeks on a HFD (figure 1A). The increased obesity in CONV-R versus GF wild-type mice was associated with increased fasting glucose and insulin levels and impaired glucose and insulin tolerance (figure 1B–F). In the absence of intact FXR signalling, the gut microbiota did not affect weight gain (figure 1A). However, the presence of gut microbiota in Fxr-deficient mice resulted in increased fasting glucose levels and impaired oral glucose tolerance; these changes were similar to the microbiota-induced changes observed in wild-type mice (figure 1B, C). By contrast, the presence of gut microbiota did not affect fasting insulin, insulin release after glucose administration nor insulin tolerance in Fxr-deficient mice (figure 1D–F). Insulin staining of tissue sections from the pancreas demonstrated that islet size and beta-cell mass were greater in CONV-R versus GF wild-type mice but were not affected by the presence of a microbiota in Fxr−/− mice (figure 1G–I).
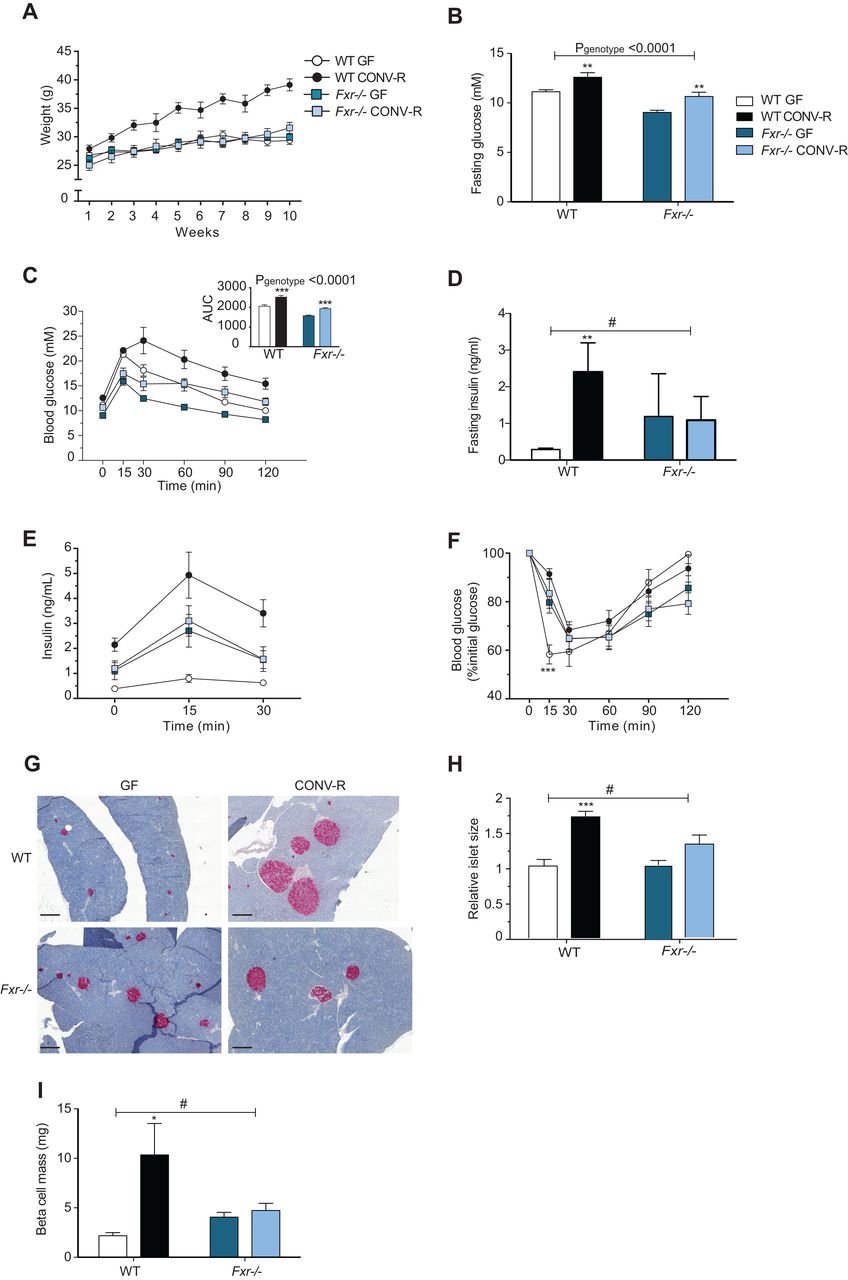
Farnesoid X receptor (FXR) and the gut microbiota regulate development of diet-induced obesity. (A) Weight gain of 9-week to 14-week old germ-free (GF) and CONV-R wild-type and Fxr−/− male mice on a high-fat diet (HFD) for 10 weeks (n=7–14 mice per group). (B) Fasting glucose levels, (C) oral glucose tolerance test, (D) fasting insulin levels and (E) blood insulin levels during the first 30 min of the oral glucose tolerance test in mice after 10 weeks on a HFD (n=6–7 mice per group). (F) Insulin tolerance test in mice after 10 weeks on a HFD (n=10–11 mice per group). (G) Pancreatic sections stained for insulin (red) and haematoxylin (blue) from mice after 10 weeks on a HFD. Scale bars, 200 μm. (H) Islet size (relative to GF wild-type) in mice after 10 weeks of HFD (n=5–9 mice per group). (I) Beta-cell mass in mice after 10 weeks on a HFD (n=5–8 mice per group). Mean values±SEM are plotted; *p<0.05, **p<0.01, ***p<0.001 versus GF mice of same genotype; #significant for genotype–colonisation interaction.
We noted that the gut microbiota did not significantly affect FXR expression in the small intestine and liver, tissues where FXR is most abundantly expressed, or in WAT (data not shown).
As expected, we found that obese CONV-R wild-type mice had increased adiposity and larger adipocytes compared with GF counterparts after 10 weeks on a HFD; this difference was abolished in Fxr−/− mice (figure 2A). In agreement, we showed that the number of crown-like structures (which indicate accumulation of macrophages around necrotic adipocytes) and the expression of the macrophage markers Emr1 (which encodes the protein F4/80), Saa3 and Tnfa (which encode proinflammatory cytokines) and Ccl2 (which promotes macrophage infiltration into WAT39) were markedly increased by the microbiota in adipose tissue from wild-type but not Fxr−/− mice (figure 2B–F).

Gut microbiota increases crown-like structures and expression of proinflammatory markers in white adipose tissue (WAT) through farnesoid X receptor (FXR). (A) Representative MAC-2 immunostaining of WAT from germ-free (GF) and CONV-R wild-type and Fxr−/− male mice on a high-fat diet (HFD) for 10 weeks. Scale bars, 100 μm. (B) Quantification of crown-like structures (n=6–7 mice per group). (C–F) qPCR analysis of Emr1, Saa3, Tnfα and Ccl2 expression in WAT from mice after 10 weeks on a HFD (n=4–9 mice per group). Mean values±SEM are plotted; *p<0.05, **p<0.01, ***p<0.001 versus GF of same genotype; #significant for genotype–colonisation interaction.
These data suggest that microbiota-induced obesity is associated with increased macrophage infiltration and that this process requires functional FXR signalling.
Effect of the gut microbiota on hepatic steatosis is reduced in Fxr−/− mice
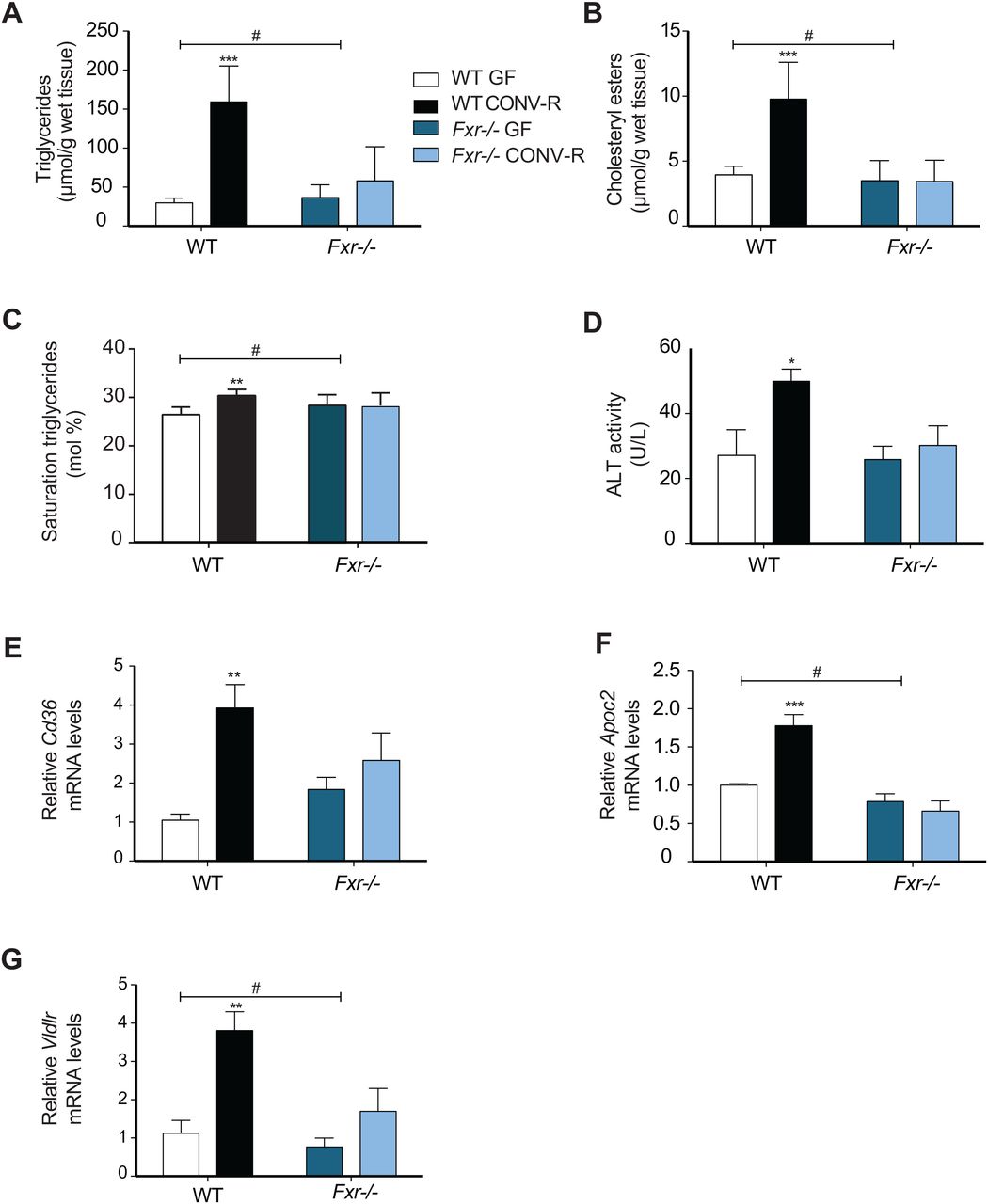
Obesity is associated with increased hepatic steatosis, which correlates with insulin resistance.40 ,41 Because FXR has been demonstrated to have a large impact on hepatic lipid metabolism,16 we investigated whether the gut microbiota induces steatosis through activating FXR signalling. Quantification using direct infusion/mass spectrometry showed higher levels of triglycerides, saturated triglycerides and cholesteryl esters in livers of CONV-R wild-type mice compared with GF mice after 10 weeks on a HFD (figure 3A–C). The increased hepatic steatosis in CONV-R wild-type mice was also associated with elevated ALT levels (figure 3D), indicating increased hepatic inflammation. By contrast, the presence of a gut microbiota did not promote hepatic steatosis or increased ALT activity in Fxr−/− mice (figure 3A–D).
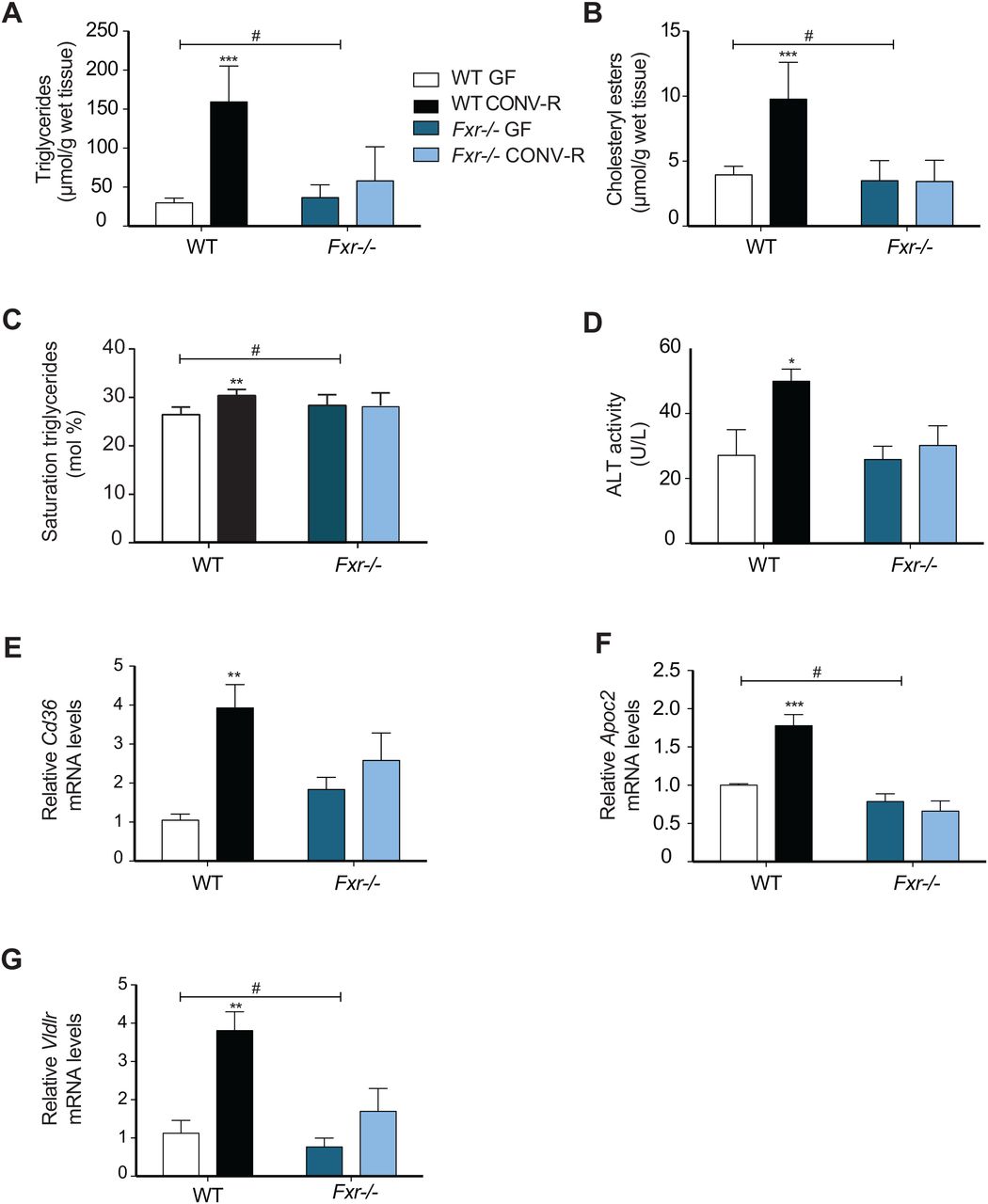
Gut microbiota increases hepatic steatosis and expression of genes involved in lipoprotein uptake through farnesoid X receptor (FXR). (A) Quantification of triglycerides, (B) saturated triglycerides and (C) cholesteryl esters in livers from germ-free (GF) and CONV-R wild-type and Fxr−/− male mice after 10 weeks on a high-fat diet (HFD) (n=4–9 mice per group). (D) Alanine transaminase (ALT) levels in the serum of mice after 10 weeks on a HFD (n=5–8 mice per group). (E–G) qPCR analysis of Cd36, Apoc2 and Vldlr expression in livers from mice after 10 weeks on a HFD (n=4–9 mice per group). Mean values±SEM are plotted; *p<0.05, **p<0.01, ***p<0.001 versus GF of same genotype; #significant for genotype–colonisation interaction.
We next investigated the effect of the microbiota on FXR-regulated genes in the liver that could potentially contribute to hepatic steatosis. The increased lipid accumulation in CONV-R wild-type mice after high-fat feeding could not be explained by increased expression of lipogenic genes such as Fas and Acc1 (see online supplementary figure S1A,B) nor by reduced expression of genes involved in fatty acid oxidation such as Ppara, Acox1 and Cpt1a (see online supplementary figure S1C–E). However, we found significant upregulation of the fatty acid transporter Cd36 as well as Apoc2 and Vldlr in liver from CONV-R versus GF wild-type mice or Fxr−/− mice with or without a microbiota (figure 3E–G). These data suggest that the microbiota may promote diet-induced hepatic steatosis at least partly through activation of FXR-regulated fatty acid and lipoprotein uptake genes in the liver.
Supplementary materials
FXR alters bile acid and gut microbiota composition
The gut microbiota and FXR have profound effects on bile acid metabolism,14–16 and here we used ultra-performance liquid chromatography–MS/MS to quantify serum and caecal bile acids from CONV-R and GF wild-type and Fxr−/− counterparts. We found that the bile acid composition in serum and caecum of GF mice was dominated by TβMCA and was not affected by Fxr genotype (see online supplementary figure S2A,B). By contrast, differences in the bile acid profiles of serum were observed when we compared CONV-R wild-type and Fxr−/− mice: CONV-R Fxr-deficient mice had a significantly higher proportion of TβMCA (see online supplementary figure S2A), suggesting reduced capacity to deconjugate bile acids. Caecal bile acids in CONV-R mice were predominantly deconjugated and dominated by secondary bile acids (see online supplementary figure S2B), as previously demonstrated in mice on chow diet.15 Consistent with the higher level of TβMCA in the serum of CONV-R Fxr−/− mice, these mice had a trend towards higher caecal levels of βMCA compared with CONV-R wild-type mice (see online supplementary figure S2B), further suggesting reduced capacity to metabolise bile acids in the absence of functional FXR signalling.
The altered serum bile acid profile was associated with reduced hepatic expression of Cyp7a1 in CONV-R wild-type mice compared with the other groups; by contrast genotype, but not microbiota, affected Cyp8b1 expression (see online supplementary figure S3A, B).
To investigate whether the metabolic differences between CONV-R wild-type and Fxr−/− mice were associated with an altered gut microbiota, we extracted genomic DNA from faecal contents of these mice and sequenced the 16S rRNA gene using MiSeq. Unweighted UniFrac analysis (qualitative; not sensitive to abundance of taxa) showed a clear separation between CONV-R wild-type and Fxr−/− mice (figure 4A). We observed decreased levels of Firmicutes and increased levels of Bacteroidetes in CONV-R Fxr−/− compared with wild-type mice after 10 weeks on a HFD (figure 4B, C). LEfSe also indicated that genera belonging to Firmicutes, Epsilon- and Gamma proteobacteria were increased in CONV-R wild-type mice and that genera belonging to Bacteroidia were increased in CONV-R Fxr−/− mice (figure 4D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Farnesoid X receptor (FXR) signalling alters the gut microbiota leading to modulation of glucose metabolism. (A) Principal coordinates analysis plot of unweighted Unifrac distances and (B–C) relative abundance of Firmicutes and Bacteroidetes in faeces of CONV-R wild-type or Fxr−/− mice on a high-fat diet (HFD) for 10 weeks (n=4–5 mice per group). (D) Cladogram generated from LEfSe analysis showing the most differentially abundant taxa enriched in microbiota from wild-type (green) and Fxr−/− (red) mice. (E) Weight gain, (F) fat content as a percentage of body weight and (G) oral glucose tolerance test in mice conventionalised with caecal microbiota from wild-type mice [CONV-D (WT)] or from Fxr−/− mice [CONV-D (Fxr−/−)] and fed a HFD for 10 weeks (n=5 mice/group). Mean values±SEM are plotted; *p<0.05, **p<0.01, ***p<0.001.
Altered gut microbiota from Fxr−/− mice modulates obesity and glucose metabolism on transfer to GF wild-type mice
Previous studies have shown that the obesity phenotype can be transferred by transplanting the microbiota from obese mice into GF recipients.3 ,10 ,11 To assess whether the altered gut microbiota of CONV-R Fxr−/− mice could contribute to the metabolic differences between CONV-R wild-type and Fxr−/− mice, we therefore colonised GF wild-type mice with the caecal microbiota from HFD-fed CONV-R Fxr−/− or wild-type mice. After 10 weeks on a HFD, mice colonised with the CONV-R Fxr−/− microbiota [CONV-D(Fxr−/−)] gained less weight than mice that were colonised with the CONV-R wild-type microbiota [CONV-D(WT)] (figure 4E). Furthermore, MRI analysis showed that adiposity was significantly lower in CONV-D(Fxr−/−) mice than in CONV-D(WT) mice (figure 4F). As we had shown that Fxr−/− mice had improved oral glucose tolerance compared with their wild-type counterparts (p<0.001; figure 1C), we investigated whether this effect could also be transferred by transplanting the microbiota. Indeed, we showed that glucose tolerance was improved in CONV-D(Fxr−/−) mice compared with CONV-D(WT) mice after 10 weeks on a HFD (figure 4G). These data suggest that the altered gut microbiota in HFD-fed CONV-R Fxr-deficient mice may at least partly contribute to some of the beneficial metabolic effects observed in these mice.
Discussion
To investigate whether the gut microbiota promotes diet-induced obesity and associated phenotypes through an FXR-dependent mechanism, we rederived Fxr-deficient mice as GF and compared them with CONV-R counterparts. We found that the gut microbiota promoted diet-induced obesity, adipose inflammation and liver steatosis in a strict FXR-dependent fashion. Furthermore, we showed that Fxr-deficient mice had an altered bile acid and gut microbiota composition. By transferring the microbiota from wild-type and Fxr−/− CONV-R mice to GF recipients, we showed that the altered microbiota partly contributed to the improved metabolic profile of Fxr-deficient mice.
As expected, lean GF wild-type mice had low fasting insulin levels and beta-cell mass, and exhibited improved insulin tolerance in comparison with obese CONV-R mice. Although the phenotypes of GF and CONV-R Fxr-deficient mice were mostly similar, highlighting microbial signalling through FXR, we noted that GF Fxr-deficient mice had improved fasting glucose and glucose tolerance compared with CONV-R Fxr−/− mice. Interestingly, the presence of a gut microbiota resulted in elevated fasting glucose levels without an effect on fasting insulin levels in Fxr-deficient mice. Although this could potentially be explained by CONV-R Fxr−/− mice having a higher degree of insulin resistance than GF Fxr−/− mice, this did not translate into impaired insulin tolerance in CONV-R Fxr−/− mice. However, it should be noted that an insulin tolerance test is mainly a reflection of overall insulin sensitivity (ie, muscle glucose usage), and cannot take into account hepatic insulin resistance. Thus, one explanation of the discrepancy between glycaemic and insulinaemic profiles could be linked to hepatic insulin resistance.
Our observation that the gut microbiota induced liver steatosis in response to a HFD in an FXR-dependent manner is in agreement with a recent finding showing that antibiotic treatment reduces steatosis through FXR signalling.24 We also observed increased expression of Cd36, Apoc2 and Vldlr in liver from wild-type mice but not Fxr-deficient counterparts. All three genes are associated with lipoprotein uptake,42–44 which may explain increased steatosis, which could not be attributed to increased expression of lipogenic genes or reduced expression of genes involved in fatty acid oxidation. Furthermore, Vldlr−/− mice have been shown to be protected against developing steatosis45 and diet-induced obesity.46
It is well known that the gut microbiota affects bile acid metabolism,15 ,23 and changes in the bile acid profile may partly explain the increased adiposity in CONV-R versus GF wild-type mice. In the absence of the microbiota, the serum bile acid profile was not affected by Fxr genotype and was dominated by TβMCA, highlighting the importance of the microbiota to promote bile acid diversity. By contrast, in the presence of a microbiota, Fxr deficiency had profound effects on the serum bile acid profile, which may be a consequence of altered gut microbial communities and/or increased production of bile acids. Bile acids can directly modulate bacterial growth and microbial ecology in the gut.47 ,48 In addition, bile acids can indirectly modulate the gut microbiota by promoting FXR-induced expression of genes producing antimicrobial agents such as Nos2 in the small intestine.49 Thus, an altered bile acid profile may potentially contribute to the alterations in host metabolism observed in Fxr-deficient versus wild-type mice by modifying the microbiota composition. To test this hypothesis, we compared the gut microbiota of wild-type and Fxr-deficient mice. We showed that the gut microbiota of Fxr-deficient mice was characterised by a phylum-wide increase in Bacteroidetes and phylum-wide reduction of Firmicutes. Similar alterations have previously been associated with protection against obesity.1 ,3 Importantly, we also showed that the transfer of the microbiota from HFD-fed Fxr-deficient mice to GF wild-type mice resulted in less fat mass gain and improved glucose metabolism compared with mice that were colonised with microbiota from HFD-fed wild-type mice. Thus, our findings suggest that the altered gut microbiota in HFD-fed Fxr-deficient mice may directly contribute to the lean phenotype.
In conclusion, our results support the concept that the gut microbiota promotes diet-induced obesity and associated phenotypes through effects on the bile acid profile and altered FXR signalling. Furthermore, our findings indicate that FXR may also contribute to increased adiposity by shifting the gut microbiota to a more obesogenic configuration. Thus, manipulation of the gut microbiota and/or FXR signalling may reveal novel ways to treat metabolic disease. However, the precise role of FXR in host metabolism is complex, and conflicting studies have reported that intestinal FXR signalling promotes23 or protects against50 obesity. Taken together, although it is clear that the gut microbiota and FXR regulate host metabolism, the underlying molecular mechanisms require further investigation.
Acknowledgments
We thank Carina Arvidsson and Anna Hallén for superb technical assistance, the genomics core facility at University of Gothenburg for 16S sequencing, Valentina Tremaroli and Rozita Akrami for assisting with microbiota analysis and Frank Gonzalez for providing Fxr−/− mice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online figures
- Data supplement 2 - Online table 1
- Data supplement 3 - Online table 2
- Data supplement 4 - Online table 3
Footnotes
↵AP and NS contributed equally.
Contributors FB conceived and designed the study; AP, NS, FS, RC, AM, MS and TG performed experiments; AP, NS, FS, RP and FB analysed and interpreted the data; AP, NS, RP and FB wrote the paper; All authors commented and approved the final manuscript.
Funding This work was supported by the Human Frontier of Science Program (RGY64/2008), Swedish Research Council, NovoNordisk Foundation, Torsten Söderberg's Foundation, Ragnar Söderberg's Foundation, Swedish Diabetes Foundation, Swedish Heart Lung Foundation, IngaBritt och Arne Lundbergs Foundation, Knut and Alice Wallenberg Foundation, Swedish Foundation for Strategic Research, the EU-funded project ETHERPATHS (FP7-KBBE-222639, www.etherpaths.org), Åke Wiberg, Magnus Bergvall, Lars Hierta's, Nanna Svartz, Fredrik and Ingrid Thurings Foundations and LUA-ALF grants from Sahlgrenska University Hospital. FB is a recipient of ERC consolidator Grant 2013 (European Research Council, Consolidator grant 615362-METABASE).
Competing interests FB is the founder and share holder of Metabogen AB.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Sequencing data will be made available in public databases.