Article Text

Abstract
Objective Patients infected with Helicobacter pylori develop chronic gastritis with a subgroup progressing to further complications. The role of microbiota from the oral cavity swallowed with saliva and either transiting the stomach or persisting in the gastric mucosa is uncertain. It is also not known whether the bacterial community differs in luminal and mucosal niches. A key question is whether H. pylori influences the bacterial communities of gastroduodenal niches.
Design Saliva, gastric and duodenal aspirates as well as gastric and duodenal biopsies were collected during oesophagogastroduodenoscopy from 24 patients (m:9, f:15, mean age 52.2±SD 14.5 years). RNA was extracted and the V1–V2 region of the retrotranscribed bacterial 16S rRNA amplified and sequenced on the Illumina MiSeq platform.
Results Overall, 687 bacterial phylotypes that belonged to 95 genera and 11 phyla were observed. Each individual comprised a unique microbiota composition that was consistent across the different niches. However, the stomach fluid enriched for specific microbiota components. Helicobacter spp were shown to dominate the mucosa-associated community in the stomach, and to significantly influence duodenal and oral communities.
Conclusions The detailed analysis of the active global bacterial communities from the five distinct sites of the upper GI tract allowed for the first time the differentiation between host effects and the influence of sampling region on the bacterial community. The influence of Helicobacter spp on the global community structures is striking.
- BACTERIAL INTERACTIONS
- HELICOBACTER PYLORI
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
The upper GI tract is inhabited by a complex microbiota.
The oral cavity may be the source of the gastric microbiota.
The presence of Helicobacter pylori diminishes species richness and may further influence bacterial community structure.
What are the new findings?
Using reverse transcribed 16S rRNA as the amplification template, precise insights into the metabolically active bacteria of the upper GI tract were obtained.
Each subject has their own individual microbiota composition that is consistent throughout the regions of the upper GI tract.
The stomach fluid enriches for specific microbiota components.
The presence of H. pylori influences the community composition of the duodenum and oral cavity.
How might it impact on clinical practice in the foreseeable future?
The bacterial community of the oral cavity may play a crucial role in determining susceptibility to H. pylori infection and must be considered in further analyses.
Stomach aspirate microbiota are very distinct from biopsies and do not provide a reliable depiction of the mucosal associated bacteria.
Changes of the microbiota in the duodenum due to H. pylori infection may influence duodenal diseases.
Introduction
Until the discovery of Helicobacter pylori, the human stomach was considered to be a sterile site due to its highly acidic nature. Following this discovery, it was assumed that H. pylori was the only bacterium able to colonise the gastric epithelium.1 However, in the proceeding years, members of the phyla Firmicutes, Proteobacteria and Bacteroidetes were cultured from gastric juice2 with more recent culture-independent work revealing a complex bacterial community inhabiting the upper GI tract.3
H. pylori is usually acquired during childhood4 ,5 is persistent throughout life and associated with various degrees of gastric mucosal inflammation.4 Infected individuals develop chronic gastritis and a subgroup progresses to further complications such as peptic ulcer disease or cancer.6 H. pylori gastritis leads to changes in acid secretion of varying degrees depending on the topographical and phenotypical expression of gastritis.6 ,7
Recent studies have indicated saliva as a source for bacteria present in stomach aspirates (SAs).8 ,9 However, there is a lack of knowledge on whether these bacteria are able to colonise the gastric mucosa. The gastric mucosa is covered by a viscous mucus layer with a gradient between the physiological acidic luminal and the neutral epithelial pH. H. pylori uses this pH gradient to colonise its ecological niche on the gastric epithelium. This difference in pH is assumed to result in differences in the transient luminal and the mucosa-associated microbiota where recent analyses using limited numbers of individuals have shown differences in the phylogenetic composition of bacterial communities.10 There is also limited knowledge on the biodiversity in the gastric mucosa of individuals infected with H. pylori compared with non-infected individuals even though it has been suggested that the carcinogenic role of H. pylori is partly due to its impact on the gastric commensal microbiota.5 ,11 However, H. pylori obviously dominates in the stomach mucosa of infected individuals while uninfected individuals show higher biodiversity.12 ,13 Changes of the microbiota in the duodenum and the proximal small bowel due to H. pylori infection have yet to be studied despite the causal relationship between a gastric infection with H. pylori and duodenal ulcer disease.14 ,15
In this study we aimed to characterise non-H. pylori bacteria, to identify whether they are transients or residents of the intestinal tract and to assess to which extent H. pylori affects the biodiversity of the whole human upper GI tract. Thus, the microbiota of saliva, SA, stomach biopsies (SBs), duodenum aspirates (DAs) and duodenum biopsies (DBs) of 24 individuals were analysed by high-throughput sequencing. Most sequence-based studies of bacterial communities have used libraries constructed by amplifying DNA extracted from samples, which include DNA from active or viable cells, and from dead lysed or degraded cells. Due to the short life span of extracellular RNA compared with DNA, sequence libraries constructed from reverse transcripts of RNA describe the active component of a bacterial community.16 ,17 Thus, the biodiversity of GI samples was analysed based on the RNA profiles.
Materials and methods
Study cohort
Individuals undergoing scheduled oesophagogastroduodenoscopy at the University of Magdeburg were enrolled between October 2014 and March 2015. The protocol was performed in accordance with current good clinical practice guidelines, the declaration of Helsinki, and was approved by the local ethics committee. All individuals gave their written informed consent. The cohort of 24 individuals with histopathological diagnosis of chronic gastritis comprised 9 men and 15 women with a mean age of 52±14 years (see online supplementary table S1). Individuals had not received antibiotic treatment for at least 4 weeks prior to sampling. From each patient, samples from saliva as well as both stomach and duodenal aspirates and biopsies were obtained. H. pylori status was identified based on the rapid urease test and histopathological assessment (see online supplementary materials). In addition, serum samples were analysed to detect anti-H. pylori IgG (see online supplementary materials).
supplementary materials
supplementary table
RNA extraction, cDNA synthesis and library preparation
RNA was extracted using the RNeasy kit (Qiagen) following manufacturer's instructions, but including a mechanical lysis step as detailed in the online supplementary materials. After DNA digestion, first-strand cDNA was synthesised using SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, California, USA) and random primers, following manufacturer’s instructions. Amplicon libraries were generated as previously described18 where the V1-V2 region of the 16S rRNA was amplified after 20 cycles PCR reaction using the 27F and 338R primers19 and sequenced on a MiSeq (2×250 bp, Illumina, Hayward, California, USA).
Bioinformatic and statistical analysis
Bioinformatic processing was performed as previously described.18 Raw reads were merged with the Ribosomal Database Project (RDP) assembler.20 Overall 3 677 068 paired-ends reads were obtained with a mean of 30 900±16 467 reads per sample. Sequences were aligned within MOTHUR (gotoh algorithm using the SILVA reference database) and subjected to preclustering (diffs=2)21 yielding so-called phylotypes that were filtered for an average abundance of ≥0.001% and a sequence length ≥250 bp before analysis. All samples were resampled to equal the smallest library size of 10 217 reads using the phyloseq package22 returning 687 phylotypes (see online supplementary table S2). Phylotypes were assigned to a taxonomic affiliation based on the naïve Bayesian classification23 with a pseudo-bootstrap threshold of 80%. Phylotypes were then manually analysed against the RDP database using the Seqmatch function as well as against the National Center for Biotechnology Information (NCBI) database to define the discriminatory power of each sequence read. A species name was assigned to a phylotype when only 16S rRNA gene fragments of previously described isolates of that species showed up to two mismatches with the respective representative sequence read.18 Similarly, a genus name was assigned to a phylotype when only 16S rRNA gene fragments of previously described isolates belonging to that genus and of 16S rRNA gene fragments originating from uncultured representatives of that genus showed up to two mismatches. Relative abundances (in percentage) of phylotypes, genera and phyla were used for downstream analyses (see online supplementary table S3–S5). The vegan package from R (Oksanen J, Guillaume-Blanchet F, Kindt R, et al vegan: Community Ecology Package. R package version 2.3-2. 2015. http://CRAN.R-project.org/package=vegan) was used to generate rarefaction curves as well as Shannon’s diversity and Pielou's evenness indices, while multivariate analyses were performed using PRIMER (V.7.0.11, PRIMER-E, Plymouth Marine Laboratory, UK), and univariate analyses using Prism 7 (Graphpad Software).
supplementary table
supplementary table
supplementary table
supplementary table
Taxonomic diversity was calculated using algorithms for taxonomic distinctness (TD): average TD (δ+) and variation in TD (λ+). δ+ represents the average taxonomic distance between all pairs of species within each sample and thus is a summary of average taxonomic breadth of each sample, while λ+ reports how consistent each level of organisation within the Linnaean classification is represented.24 ,25
The data matrices comprising either the 687 phylotypes or the 95 genera were used to construct sample-similarity matrices using the Bray-Curtis algorithm,26 where samples were ordinated using non-metric multidimensional scaling (nMDS) with 50 random restarts.27 Significant differences between a priori predefined groups of samples were evaluated using analysis of similarity (ANOSIM) (9999 permutations)27 and/or permutational multivariate analysis of variance (PERMANOVA), allowing for type III (partial) sums of squares, fixed effects sum to zero for mixed terms and Monte Carlo p values generated using unrestricted permutation of raw data.28 Groups of samples were considered significantly different if the p value was <0.05. The abundances of phyla, genera and of those phylotypes that represent ≥0.5% of the total community in at least one sample (379 phylotypes), were compared by the Mann-Whitney test with Benjamini-Hochberg correction29 for multiple comparisons. Groups of samples were considered significantly different if the adjusted p value was <0.05. For analysing the effect of H. pylori on the upper GI tract, communities of saliva (OA), SA, DA, SB and DB of individuals infected with and without H. pylori (H and X respectively), were compared: OAX/OAH, SAX/SAH, DAX/DAH, SBX/SBH, and DBX/DBH respectively. Differences in bacterial communities of aspirates versus biopsies and between biopsies were identified by comparing SAX/SBX, DAX/DBX, SBX/DBX, SBX/DAX as well as SAH/SBH, DAH/DBH and SBH/DBH. Differences across different regions were identified by comparing OAX/SAX, SAX/DAX, OAH/SAH and SAH/DAH. Differences in the abundance of genera and phylotypes in comparisons comprising SAH and DAH were performed excluding Helicobacter reads.
Factors ascribed to individuals in this study (age, gender, use of proton pump inhibitors and suffering from intestinal metaplasia or not) were used to test for differences between groups of individuals being infected with and without H. pylori using the unpaired t-test with Welch's correction or the Fisher's exact test (two-tailed, CIs at 95%) (see online supplementary table S1).
Results
Active bacterial communities present in saliva, gastric and duodenal fluids and gastric and duodenal biopsies from 24 individuals comprising 8 individuals with a clinical and histopathological diagnosis of H. pylori-induced gastritis and 16 individuals with chronic gastritis but without H. pylori infection were compared to identify individual as well as site-specific bacterial signatures and to identify the influence of H. pylori on the global community structures. Following sequencing, rarefaction analysis (see online supplementary figure S1) and rarefying of library sizes, 687 phylotypes were observed (see online supplementary table S2 and S3). The phylotypes belonged to 95 genera and 11 phyla, where sequences of Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria and Fusobacteria comprised approximately 99% of the total bacterial community (see online supplementary tables S4 and S5).
supplementary figures
Interindividual variation in the global bacterial assemblages
The global bacterial profiles at phylotype level were compared using group-average agglomerative hierarchical clustering following sample-pairwise comparisons (figure 1). Assemblages across the upper GI tract from each individual typically clustered together irrespective of sample origin. Only communities obtained from SB from H. pylori positive individuals clustered based on sample origin, due to the high abundance of H. pylori.

Group-average agglomerative hierarchical clustering of 119 samples, based on global bacterial profiles (phylotype-level) along the upper GI tract (saliva, O; stomach, S; and duodenum, D) where aspirates (A) and biopsies (B) from 24 individuals infected with (H) or without (X) Helicobacter pylori. Individuals are denoted by unique symbols. Four selected phylotypes, which were present in high abundance in samples from single individuals are shown as inserts. The y axis shows the relative abundance of the phylotype in a given sample, while the x axis shows samples originating from individual 1–24, which in each case are given in the order OA, stomach aspirates (SA), duodenum aspirates (DA), stomach biopsies (SB) and duodenum biopsies (DB).
This strong host effect at phylotype level is supported by PERMANOVA confirming a strong and statistically significant difference between individuals overall (Pseudo-F=4.46, p=0.001), where 230 of the 253 possible sample-pairwise comparisons were significantly different (p<0.05) (see online supplementary table S6). As an example of the uniqueness of samples from a single individual, the communities obtained from P5 are dominated by a single phylotype (Phy10, Streptococcus salivarius) accounting for 53% of reads obtained from duodenal aspirates, where only three further individuals contained this phylotype in an abundance >1% in any community (figure 1). P2 was the only individual, where microorganisms of the candidate phylum SR1 (Phy312) reached a high relative abundance (2.5%).
supplementary table
Similarities in the bacterial community assemblages between locations of the upper human GI tract
Formal comparisons between the global bacterial assemblages across the three different regions of the upper GI tract and between biopsies and individuals infected with or without H. pylori revealed that the global bacterial structures were indeed significantly different (PERMANOVA pseudo-F=2.71, p=0.001) as also indicated in the nMDS plot (figure 2A, B). Bacterial communities of SB and SA were significantly different in both infected and non-infected individuals as assessed by PERMANOVA and ANOSIM (see figure 2C and online supplementary table S7). Communities associated with saliva and SA of non-infected individuals showed very similar community structures (figure 2).

Differences in global bacterial community structures along the upper GI tract (saliva, O; stomach, S; and duodenum, D) of aspirates (A) and biopsies (B) from 24 individuals infected with (H) or without (X) Helicobacter pylori, as assessed by non-metric multidimensional scaling (nMDS). (A) Global community structure based on standardised phylotype abundance data. (B) Global community structure excluding stomach biopsies of H. pylori-infected individuals based on standardised genus abundance data. (C) Differences between groups of samples as evaluated using analysis of similarity (ANOSIM) with the R statistic measuring the degree of separation between groups where ‘ns’ denotes ‘not significant’.
supplementary table
Comparisons between the global bacterial assemblages at genus level (see online supplementary table S6 and figure S2) as compared with the phylotype level showed a less pronounced influence of the host as only 87 pairwise comparisons (34%) between individuals showed the presence of distinct communities, indicating that individual differences are highly governed by differences in phylotypes rather than genera. PERMANOVA analysis of bacterial assemblages across the three different regions of the human upper GI tract at genus level showed the same statistically significant differences between those regions where differences had been observed at phylotype level (see online supplementary table S7).
The phylotype richness differed only to a minor extent between the different sampled sites and a significant difference was observed only in biopsies of H. pylori-infected individuals (figure 3) where SBH samples showed a much lower phylotype diversity and phylotype evenness compared with all other samples. Neither a reduction in the phylotype richness nor evenness was observed in DBH samples (figure 3), indicating that the effect of H. pylori on community diversity is restricted to the stomach. Diversity measures of communities obtained from individuals without H. pylori infection along the upper GI tract were all similar (figure 3).
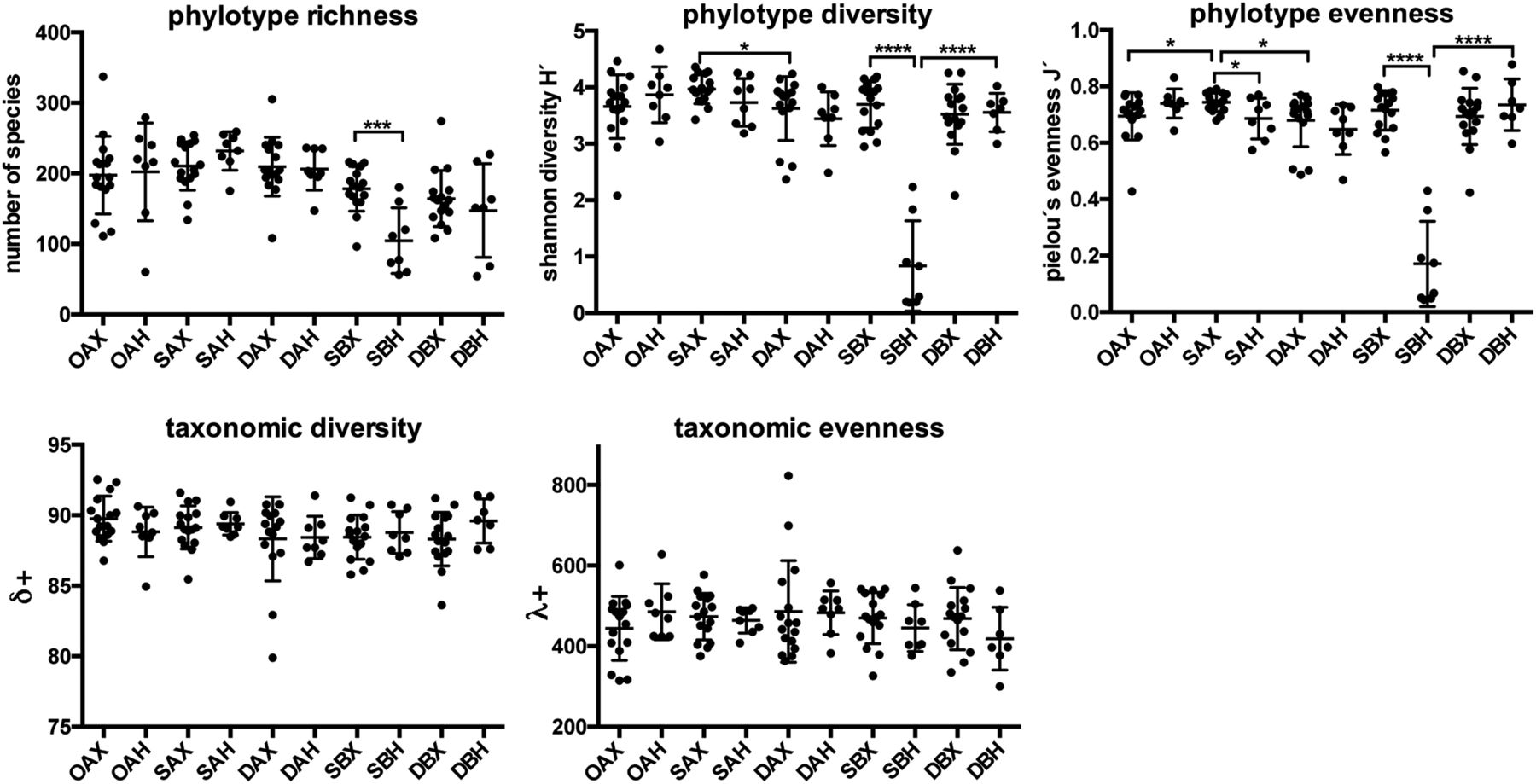
Upper GI bacterial community diversity indicated by total phylotype number, Shannon diversity (H'), Pielou's evenness (J') taxonomic diversity (δ+) and taxonomic evenness (λ+), respectively. Statistically significant differences between groups of samples from adjacent sites or from the same region but between individuals infected with or without Helicobacter pylori are indicated by ****p<0.0001; ***p<0.001. *p<0.05. The mean and SD are shown.

Differences in bacterial community structures along the upper GI tract of aspirates from individuals not infected with Helicobacter pylori. (A) Relative mean abundance of phyla and (B) relative abundance of genera predominant in oral (OAX), stomach (SAX) and duodenal aspirates (DAX) where significantly differently distributed phyla and genera are indicated with asterisks *p<0.05, **p<0.01, ***p<0.001. (C) Cladogram showing the differently distributed genera and phylotypes (*p<0.05, **p<0.01, ***p<0.001). (D) Abundance of selected phylotypes which were differently distributed between OAX, SAX and DAX. Colours of lines used in (C) and colours in (D) correspond to the colour code used for phyla given in (A).
In contrast, there were no statistically significant differences in average TD (δ+) and variation in TD (λ+) between groups of samples. Thus, while the presence of H. pylori changed the phylotype diversity and richness, it did not change the taxonomic representation (see figure 3 and online supplementary figure S3).
Bacterial community in the fluids of the upper human GI tract of individuals without H. pylori infection
Differences in bacterial community composition between sample sites were already evident at the phylum level (figure 4A). While communities of saliva (OAX) and stomach aspirates (SAX) did not differ significantly, clear differences were observed between SAX and duodenum aspirates (DAX). The relative abundance of Actinobacteria and Firmicutes was higher in DAX (p=0.03 and 0.004, respectively, online supplementary table S8), whereas the relative abundance of Bacteroidetes and Fusobacteria was lower (p=0.0018 and 0.0011, respectively). At the genus level, the upper human GI tract communities in H. pylori-negative individuals were dominated by Streptococcus and Prevotella (figure 4B). Seven genera differed in abundance between OAX and SAX, with Veillonella being more abundant in OAX and Eubacterium, Fusobacterium, Lachnoanaerobaculum, Olsenella, Parvimonas and Tessarococcus being more abundant in SAX (see figure 4C and online supplementary table S9).

Differences in bacterial community structures between stomach and duodenum biopsies and aspirates in individuals not infected with Helicobacter pylori. (A) Relative mean abundance of phyla and (B) relative mean abundance of low abundant phyla. (C) Cladogram showing the differently distributed genera and phylotypes. Colours of lines in (C) correspond to the colour code used for phyla given in (A). If a genus or phylotype is differently distributed between SAX/SBX, SAX/DAX, DAX/DBX or SBX/DAX (p>0.05), the respective genus or phylotype name is coloured, with the colour code given at the bottom of (C). The level of statistical significance is indicated above the respective colour code. (D) Relative mean abundance of prominent genera in stomach aspirates (SAX) and biopsies (SBX) as well as in duodenal aspirates (DAX) and biopsies (DBX). Significantly differently distributed phyla and genera are indicated with asterisks *p<0.05, **p<0.01 and ***p<0.001.
supplementary table
supplementary table
Differences were more pronounced when SAX and DAX samples were compared. Twenty-two genera were differently abundant where the higher abundance of Firmicutes in DAX can be attributed to a higher abundance of Streptococcus, and the lower abundance of Bacteroidetes and Fusobacteria to a lower abundance of Prevotella and Fusobacterium (see figure 4C and online supplementary table S9), respectively, among others. Different niches for different genera became apparent with various Proteobacteria, Actinobacteria and Staphylococcus being virtually absent from SAX (figure 4C).
Analysis at phylotype level revealed that Phy44 (Fusobacterium) had the significantly highest abundance in SAX (figure 4B, C). Also Phy269 (Campylobacter gracilis) was specifically abundant in SAX and other Campylobacter phylotypes showed the same trend (figure 4C, D). Of the Bacteroidetes phylotypes, those indicating the presence of Prevotella showed a higher abundance in SAX compared with DAX (figure 4C, D). In contrast, Staphylococcus phylotypes showed the lowest abundance in SAX compared with DAX (see figure 4C). A similar low abundance specifically in SAX, indicating vulnerability of the respective organism to acidic conditions, was observed for various actinobacterial and γ-proteobacterial phylotypes (see figure 4C and online supplementary table S10).
supplementary table
Bacterial communities in the fluids of the stomach and duodenum compared with biopsies
An analysis at phylum level showed a clear difference between SAX and SBX in the abundance of Fusobacteria, Bacteroidetes and Firmicutes (see figure 5A and online supplementary table S8) whereas those between DAX and DBX were restricted to Spirochaetes (figure 5B). Accordingly, differences in community composition between DAX and DBX samples at genus level showed Treponema to be significantly more abundant in DAX (figure 5C). In addition, four proteobacterial and two actinobacterial genera differed significantly in abundance. In contrast, 22 genera were differently abundant in SBX versus SAX samples and Fusobacterium strains were responsible for the higher abundance of Fusobacteria in SAX while Streptococcus, Staphylococcus and Enterococcus for the higher abundance of Firmicutes in SBX (see figure 5C, D and online supplementary table S9). Members of the genera Alloprevotellla, Porphyromonas and Tannerella were responsible for the higher abundance of Bacteroidetes in SAX whereas Bacteroides followed a different trend being more abundant in SBX (figure 5C). Both genera of higher abundance in SAX and those of higher abundance in SAX were observed among the Actinobacteria and Proteobacteria (figure 5C). Fourteen of the 22 genera detected in significantly different abundance in SBX compared with SAX were also in significantly different abundance in DAX compared with SAX (see figure 5C), which may indicate SAX to be distinct from SBX, DAX and DBX with regard to the genera distribution pattern. Distributions observed at genus level were also evident at the phylotype level (see figure 5C and online supplementary table S10).
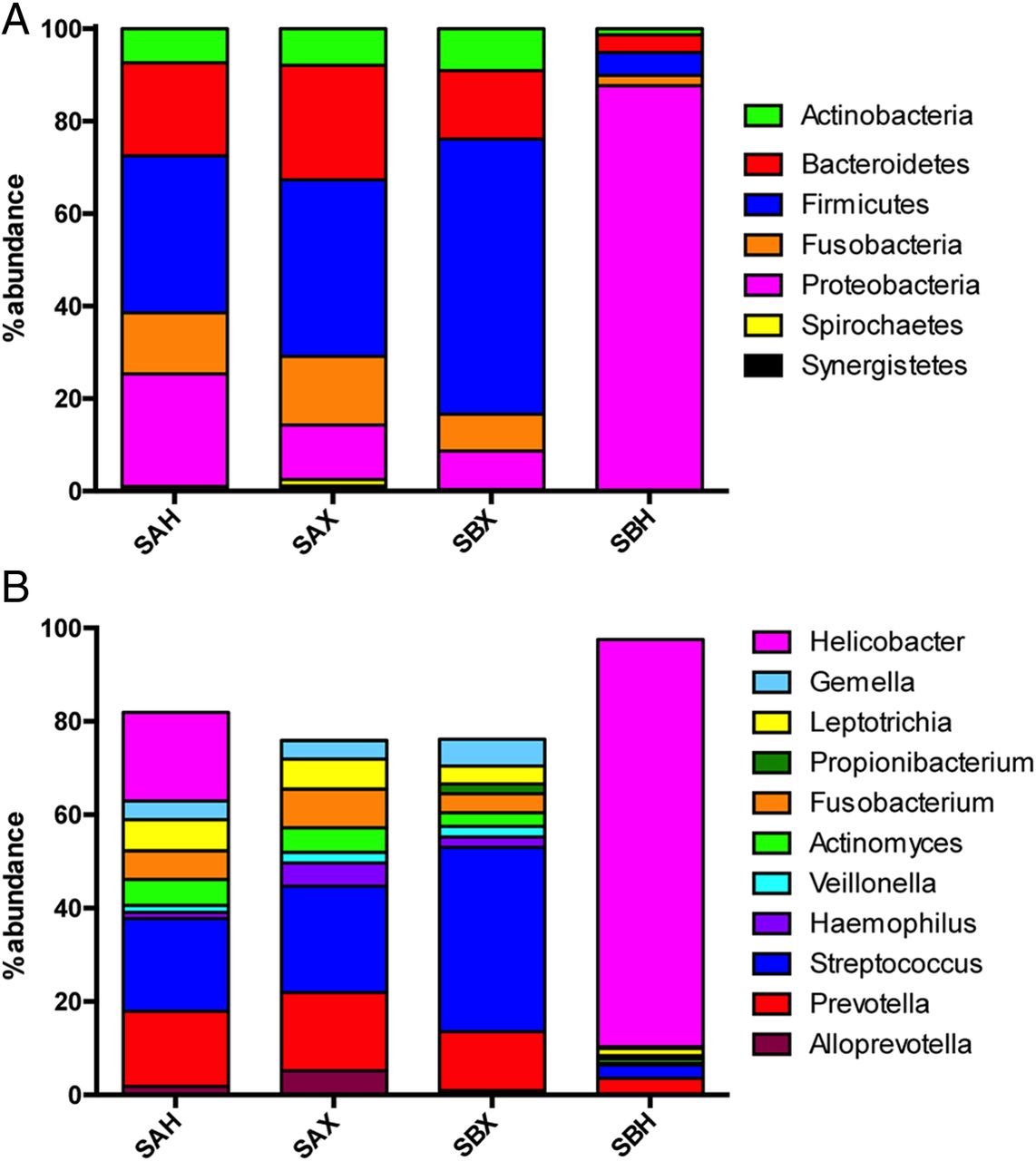
Differences in bacterial community structures between stomach aspirates and biopsies in individuals infected with and without Helicobacter pylori. (A) Relative mean abundance of phyla and (B) relative mean abundance of genera in stomach aspirates of individuals infected with (SAH) or without H. pylori (SAX) as well as in stomach biopsies of non-infected (SBX) or infected individuals (SBH).
H. pylori infection affects the stomach microbiota
Infection with H. pylori is typically manifested by a high abundance of this organism on the mucosa, and accordingly, SBH samples revealed the presence of Helicobacter in amounts exceeding 50% in all infected individuals (figure 6A). Due to the high relative abundance of H. pylori, the relative abundance of all four major phyla, that is, Actinobacteria, Bacteroidetes, Firmicutes and Fusobacteria, was low in SBH compared with SBX.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Differences in bacterial community structures between duodenum aspirates and biopsies and saliva in individuals infected with and without Helicobacter pylori. (A) Relative mean abundance of phyla and (B) relative mean abundance of genera in duodenum aspirates of individuals infected with (DAH) or without H. pylori (DAX) as well as in duodenum biopsies of non-infected (DBX) or infected individuals (DBH). (C) Abundance of those genera which were significantly differently distributed in duodenal samples between infected or non-infected individuals with asterisks denoting *p<0.05 and **p<0.01. (D) Relative mean abundance of phyla which were differently distributed between OAX and OAH samples. Colours used in C and D correspond to the colour code used for phyla given in (A).
Three different H. pylori phylotypes were dominant in SBs with Phy1 dominating in six individuals and Phy27 and Phy36 dominating in one individual each. The same Helicobacter phylotypes dominating in biopsies were also observed in the respective SAs, however to a much lesser extent and seven of the eight individuals were infected by only one Helicobacter phylotype.
Analysis of the community composition of SAH versus SAX revealed significant differences only in the relative abundance of Proteobacteria, which were of higher abundance in SAH, concomitant with a lower relative abundance of all nine other phyla detected in this niche (figure 6A). Similarly, at genus level, besides the increase in Helicobacter only Haemophilus decreased significantly in abundance in SAH samples (figure 6B) while at phylotype level only Phy25 (Haemophilus) and Phy80 (Campylobacter conciscus) showed a significant decrease (see online supplementary tables S9 and S10).
Influence of H. pylori on duodenal and oral communities
The only significant influence of H. pylori infection on duodenal communities at the phylum level was the increased abundance of Proteobacteria in DAH compared with DAX probably due to transfer of Helicobacter from the stomach to the duodenal lumen (figure 7A). Infection with H. pylori may have resulted in a lower abundance of Firmicutes and a higher abundance of Bacteroidetes in DBH biopsies of infected compared with DBX biopsies of uninfected individuals, however, the differences were statistically not significant. Except for the different abundances of Helicobacter in aspirates, no statistically significant differences were observed at genus level (figure 7B). Differences became apparent at phylotype level. The differences in biopsies seem to be minor and only Phy78 (Rhodobacteriaceae) and Phy269 (Campylobacter gracilis) were significantly reduced in abundance in biopsies (p=0.05, figure 7C). Other phylotypes were only observed in biopsies of non-infected individuals (Phy140, Streptococcus infantis; Phy168, Actinomyces; Phy81, Enterococcus and Phy478, Lachnospiraceae), indicating some influence of H. pylori. Besides Phy1 (H. pylori), which was observed in high abundance in the duodenal aspirates of infected individuals, only Phy103 (Staphylococcus aureus) was observed in significantly different abundances in DAH versus DAX samples, being more abundant in DAX (see figure 7C and online supplementary table S10).
There was no significant difference in oral communities between individuals infected with and without H. pylori, neither at phylum (figure 7A) nor at genus level. However, of the phylotypes three were significantly more abundant in non-infected individuals (Phy18, Propionibacterium acnes; Phy25, Haemophilus and Phy62, Prevotella oris) while Phy475 (Treponema) was absent from all non-infected individuals but observed in four of the eight infected individuals (figure 7D).
Discussion
Despite the high number of studies addressing the bacterial biodiversity of the human upper GI tract,30 there is still a lack of knowledge regarding the role of the commensal bacterial community in this niche. In this study, the bacterial communities of five sites from 24 individuals were defined where the use of reverse-transcribed 16S rRNA instead of 16S rDNA as template allowed the detection of the metabolically active component of the community.16 ,17
Previous studies on the gastric bacterial communities of biopsies or aspirates have not permitted the characterisation of different ecological niches in their anatomical continuity.3 ,10–12 By analysing throat biopsies and SBs from three individuals each it was reported that prominent genera of the stomach were also abundant in the throat and may be swallowed microorganisms.12 Further studies have compared the saliva and gastric microbiome of individuals characterised by an exceptional abundance of Enterobacteriaceae9 or were biased by the inclusion of individuals infected with H. pylori, who dominated gastric mucosal communities31 allowing only very limited conclusions. The identification of host specific phylotypes in our study evidences the saliva as the main source for the gastric microbiome. In fact, each subject has an indigenous microbiota composition, which is consistent throughout the investigated regions that are connected by a constant fluid drain. This underlines the concept of a continuous bacterial migration through the upper GI tract with the oral cavity as the dominant source of active bacteria. Our analyses of the active microbiota of the stomach confirm that Veillonella is depleted and Fusobacterium enriched in aspirates compared with saliva. Various groups of organisms were depleted in SA, for example Propionibacterium. This shows the analysis of the active microbiota to be advantageous compared with the analysis of bacterial presence8 and indicates the stomach environment to select against various organisms, which are active in either saliva or in the duodenum such as Staphylococcus or Streptococcus.
Despite the considerable variation in the gastric microbiota composition between individuals and also between studies, studies usually state Streptococcus, Lactobacillus, Bacteroides, Staphylococcus, Prevotella, Fusobacterium and Veillonella among others3 ,10 ,32 as prominent genera. Information on intraindividual differences between luminal and mucosal samples, however is scarce2 ,10 ,13 and studies were usually performed on a small number of individuals and biased by the inclusion of individuals infected with H. pylori. In this work, the mucosal samples comprised relatively lower concentrations of phylotypes belonging to the Bacteroidetes genera Prevotella, Alloprevotella, Porphyromonas and Tannerella, and relatively higher concentrations of phylotypes belonging to the Firmicutes genera Streptococcus and Staphylococcus as well as the Actinobacteria genera Corynebacterium, Kocuria and Propionibacterium. This provides evidence for a major difference between the luminal community, probably adapted to a low pH value and the mucosa adherent community of the stomach.
In the duodenum, Streptococcus and Prevotella were described as the most prominent genera.33 However, a recent sequencing analysis on five healthy subjects indicated Prevotella and generally Bacteroidetes to be virtually absent from duodenum mucosa.34 More recently the Proteobacteria genera Acinetobacter, Neisseria and Haemophilus, and Prevotella were reported to be the most predominant genera.35 The observation here of Streptococcus, Prevotella, Actinomyces and also of Propionibacterium, Gemella and Fusobacterium as abundant in DB is based on a cohort of 24 individuals and thus gives, for the first time, a representative overview of this environment. Only minor differences between the mucosal communities were evident, which is in strong contrast to the clear preference of Helicobacter to colonise the stomach mucosa. More generally, Campylobacter phylotyopes were preferentially found in aspirates and Enhydrobacter in mucosal samples. Similarly, only minor differences were noted between duodenal aspirate and mucosal communities. One recent study had also compared the communities of duodenal and luminal biopsies in nine volunteers noting that Acinetobacter, Bacteroides and Prevotella were the most abundant genera in biopsies, and Prevotella, Stenotrophomonas and Streptococcus the most abundant ones of the lumen.36 Stenotrophomonas was absent from all active communities analysed here and can be assumed to be present only in exceptional cases.
In accordance with previous reports,3 ,13 ,31 the gastric mucosa of Helicobacter-infected individuals was highly dominated by this organism concomitant with the relative depletion of other genera. Even though the presence of H. pylori had a significant influence on species’ richness and evenness, it did not change the taxonomic diversity. Interestingly, a recent report37 claimed that H. pylori-infected adults are likely to have higher abundances of Spirochaetes, Acidobacteria and non-Helicobacter Proteobacteria, of which none were detected in high abundance in this study. Strikingly, the influence of Helicobacter on the community was more evident in the duodenal samples and respective influences have, to our knowledge, yet to be reported. Duodenal ulcers and gastric cancer as complications of H. pylori infection have been reported to coexist in a small number of patients38 and it is assumed that undetected factors such as different compositions of the bacterial community beyond H. pylori contribute to this pathology.
Interestingly, infection with H. pylori influences the composition of the oral bacterial community and/or vice versa. Assuming an oral to oral transmission route of H. pylori 4 differences in the active oral community might influence the susceptibility of the host to H. pylori infection and a better understanding on the interaction between H. pylori and the oral community might give insights into the mode of transmission.
Acknowledgments
The authors thank Iris Plumeier and Silke Kahl for technical assistance, Robert Geffers and Michael Jarek for sequencing support, and Diego Chaves-Moreno and Robert Thänert for critical discussions.
References
Footnotes
Contributors DHP, PM and CS contributed to the study design, analysis and interpretation of the data. DHP and CS drafted the manuscript. CS, KS and PM collected samples and identified suitable subjects. DHP and PM supervised the study procedures. CS, NK and APAO established analyses and performed laboratory workup. MV, DHP, MLW-O and RV-V performed bioinformatic and statistical analyses. All authors read and approved the final version of the manuscript.
Funding This study was supported by iMed, the Helmholtz Association's Initiative on Personalized Medicine. CS was supported by CRC854, a research programme by the German funding organisation DFG.
Competing interests None.
Ethics approval University of Magdeburg operation number 31/11.
Provenance and peer review Not commissioned; externally peer reviewed.