Article Text

Abstract
Objective Diverticular disease is a common complex disorder characterised by mucosal outpouchings of the colonic wall that manifests through complications such as diverticulitis, perforation and bleeding. We report the to date largest genome-wide association study (GWAS) to identify genetic risk factors for diverticular disease.
Design Discovery GWAS analysis was performed on UK Biobank imputed genotypes using 31 964 cases and 419 135 controls of European descent. Associations were replicated in a European sample of 3893 cases and 2829 diverticula-free controls and evaluated for risk contribution to diverticulitis and uncomplicated diverticulosis. Transcripts at top 20 replicating loci were analysed by real-time quatitative PCR in preparations of the mucosal, submucosal and muscular layer of colon. The localisation of expressed protein at selected loci was investigated by immunohistochemistry.
Results We discovered 48 risk loci, of which 12 are novel, with genome-wide significance and consistent OR in the replication sample. Nominal replication (p<0.05) was observed for 27 loci, and additional 8 in meta-analysis with a population-based cohort. The most significant novel risk variant rs9960286 is located near CTAGE1 with a p value of 2.3×10−10 and 0.002 (ORallelic=1.14 (95% CI 1.05 to 1.24)) in the replication analysis. Four loci showed stronger effects for diverticulitis, PHGR1 (OR 1.32, 95% CI 1.12 to 1.56), FAM155A-2 (OR 1.21, 95% CI 1.04 to 1.42), CALCB (OR 1.17, 95% CI 1.03 to 1.33) and S100A10 (OR 1.17, 95% CI 1.03 to 1.33).
Conclusion In silico analyses point to diverticulosis primarily as a disorder of intestinal neuromuscular function and of impaired connective fibre support, while an additional diverticulitis risk might be conferred by epithelial dysfunction.
- diverticular disease
- intestinal motility
- genetic polymorphisms
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Diverticular disease is among the most common diseases of the GI tract.
Up to 2018, only three loci ( ARHGAP15, FAM155A, COLQ) of genome-wide significance had been reported.
Recently, a replication analysis of a UK Biobank genome-wide association study (GWAS) by Maguire et al identified 37 additional susceptibility loci with genome-wide significance and a replication of 8 of these loci in a Michigan population cohort.
Significance of this study
What are the new findings?
Here, we report the to date largest and most detailed GWAS with a sample size of 451 099 individuals to identify genetic risk factors for diverticular disease.
We report 48 loci with genome-wide significance, of which 12 are novel.
We were able to replicate 27 of these loci in specifically recruited replication samples from a GI specialty service with colonoscopy data available in all controls.
In addition, we replicated further eight risk loci in a combined meta-analysis with data from a Michigan population cohort.
The current study increases the number of replicated susceptibility loci for diverticular disease to 35, of which 25 loci had previously not been replicated.
Results point to diverticular disease primarily as a disorder of intestinal neuromuscular function, impaired mesenteric vascular smooth muscle function and of impaired connective fibre support.
The diverticulitis risk might be conferred by epithelial dysfunction.
How might it impact on clinical practice in the foreseeable future?
The results from this GWAS provide deep new insights into the colonic biology and disease pathophysiology of diverticular disease.
Diverticular disease is a common complex disorder characterised by mucosal outpouchings of the colonic wall at sites of relative weakness in the muscle layers close to penetrating blood vessels.1 2 The incidence of diverticular disease has increased to 50% for individuals older than 60 years and a significant rise of incidence and hospitalisation rates has been seen in younger age groups.3 Although the majority of patients harbouring diverticula remain asymptomatic throughout life, 10%–25%4–8 experience complications such as acute diverticulitis, abscess, fistula formation, bleeding or perforation. These complications cause an annual mortality of ~1 per 100 0009 due to the need for inpatient treatment and sigmoid resection after repeated episodes of diverticulitis. Owing to its high prevalence and associated complications, diverticular disease is the fifth most costly GI disease in Western countries.10
The pathogenesis of diverticular disease is thought to be a multifactorial process that involves lifestyle factors (smoking, physical inactivity, high body mass index (BMI)), structural and functional changes of the colonic wall, ageing and a genetic predisposition.11 In contrast to its high clinical and economic impact, diverticular disease is under-researched in terms of its pathophysiology.1 Epidemiological12 and twin studies13 have estimated the heritability of diverticular disease at 40%–53%. A previous genome-wide association study (GWAS) from Iceland identified associations of variants in ARHGAP15 and COLQ with uncomplicated diverticular disease and variants in FAM155A with diverticulitis.14 Additionally, 37 susceptibility loci with genome-wide significance were identified in a recent study from Maguire et al,15 with replication of 8 loci.
We report a total of 48 risk loci with genome-wide significance and consistent OR in a replication sample of 3893 cases and 2829 diverticula-free controls as verified by colonoscopy. We were able to replicate 27 of these loci in specifically recruited replication samples from a GI specialty service with colonoscopy data available in all controls. The large number of loci we identified and our functional follow-up provide novel insight into the pathophysiology of diverticular disease as a disorder of intestinal neuromuscular function, vascular smooth muscle function and impaired connective fibre support.
Patients and methods
Study participants
An individual was classified as a diverticular disease case if they matched hospital-based International Classification of Diseases (ICD)-9 or ICD-10 coding (562, K57) in the UK Biobank dataset (n=31 964). Control individuals were classified on the basis of absence of a diverticular disease diagnosis (n=419 135). Depth of ICD coding was insufficient to differentiate disease subtype diverticulosis (ie, diverticular disease without inflammation) from diverticulitis in the UK Biobank dataset. Replication samples were obtained from Germany, Austria, Lithuania and Sweden from GI specialty services. Details of recruitment and phenotype ascertainment for diverticulosis and diverticulitis for each cohort are described in the online supplementary materials and methods section. An overview of the study population is provided in table 1.
Supplemental material
Study populations
GWAS analysis
Discovery GWAS analysis was performed on UK Biobank on V.3 imputed genotypes using BOLT-LMM V.2.34, which applies a linear mixed model to adjust for the effects of population structure and individual relatedness.16 This enabled the inclusion of all related individuals in our white European subset allowing a sample size of 451 099 individuals as detailed in online supplementary materials and methods.
Loci discovery and functional annotation
Genomic risk loci, lead variants and candidate single nucleotide polymorphism (SNPs) were derived from FUnctional Mapping and Annotation of genetic associations (FUMA V.1.3.1)17 based on GWAS summary statistics. Candidate SNP and gene positions are provided in online supplementary table 1 and 2. Functional consequences were assessed using ANNOVAR, a tissue-specific cis-eQTL dataset (GTExV7, https://gtexportal.org) and 15-core chromatin states (ENCODE, 2012) as detailed in the online supplementary materials and methods section.
Supplemental material
Annotation of candidate genes
In order to identify candidate gene(s) at the respective genomic risk locus, we followed i) a manually curated selection process based on local linkage disequilibrium (LD) structure and supporting evidence from regulatory elements (eQTL and chromatin interaction), outlined in online supplementary table 3 and ii) we performed hypothesis-free functional and gene annotations based on the genomic positions of risk loci using FUMA,17 as the manually curated selection process of candidate genes might not capture the full biology of the risk architecture, as detailed in online supplementary materials and methods section.
Replication genotyping and meta-analysis
Top GWAS-associated loci (n=51; p<5×10−8) were validated in a combined European sample of 3893 cases and 2829 diverticula-free controls based on colonoscopy (table 1) using the most significant discovery variant or appropriate proxies when direct genotyping of a lead variant was not technically feasible. Logistic regression analyses were performed with PLINK,18 cohort-specific β effect estimates were combined with META.19 For replication a nominal significance level of p<0.05 and consistency in OR direction between the discovery and replication stage was applied. Additional replication was achieved by including replication data presented by Maguire et al 15 (online supplementary table 4) from European samples (n=29 367) from the Michigan genome initiative into a combined meta-analysis of all European replication cohorts (n=36 089 samples). Details on the genotyping, quality control and meta-analysis are provided in the online supplementary materials and methods section.
GWAS and replication results: newly discovered and novel replicated risk loci
GWAS and replication results: confirmed, previously replicated risk loci and currently not replicated risk loci
mRNA expression analysis and immunohistochemistry
Colonic tissue samples were obtained during surgical resection. Characteristics of patients used for RT-PCR are provided in online supplementary table 7. RT-primer sequences are provided in online supplementary table 8. Layer-specific and disease-specific expression analysis results are shown in online supplementary table 9 and 10. Fluorescence immunohistochemistry was performed as previously described.20 Details on sample processing are provided in the online supplementary materials and methods section.
Gene set and pathway analysis
We used two gene set and pathway analysis approaches (MSigDB21 and VEGAS2pathway22) to determine if the polygenic signal measured in the diverticular disease associated genes clustered in specific biological pathways. Lead candidate genes (tables 2 and 3) were tested for over-representation with gene sets curated in MSigDB6.1. Results are provided in online supplementary table 11 and 12. VEGAS2pathway results are provided in online supplementary table 13 and 14.
Enrichment analyses in cell lines and primary tissues
We used GARFIELD to identify significant enrichment patterns in our GWAS findings with regulatory or functional annotations in cell lines and primary tissue derived from ENCODE and Roadmap epigenomics data (online supplementary table 15). GWAS SNPs were pruned (LD r2>0.1) and then annotated based on functional information overlap. Further details are provided in the online supplementary materials and methods section.
Results
Genome-wide association study and validation of the loci
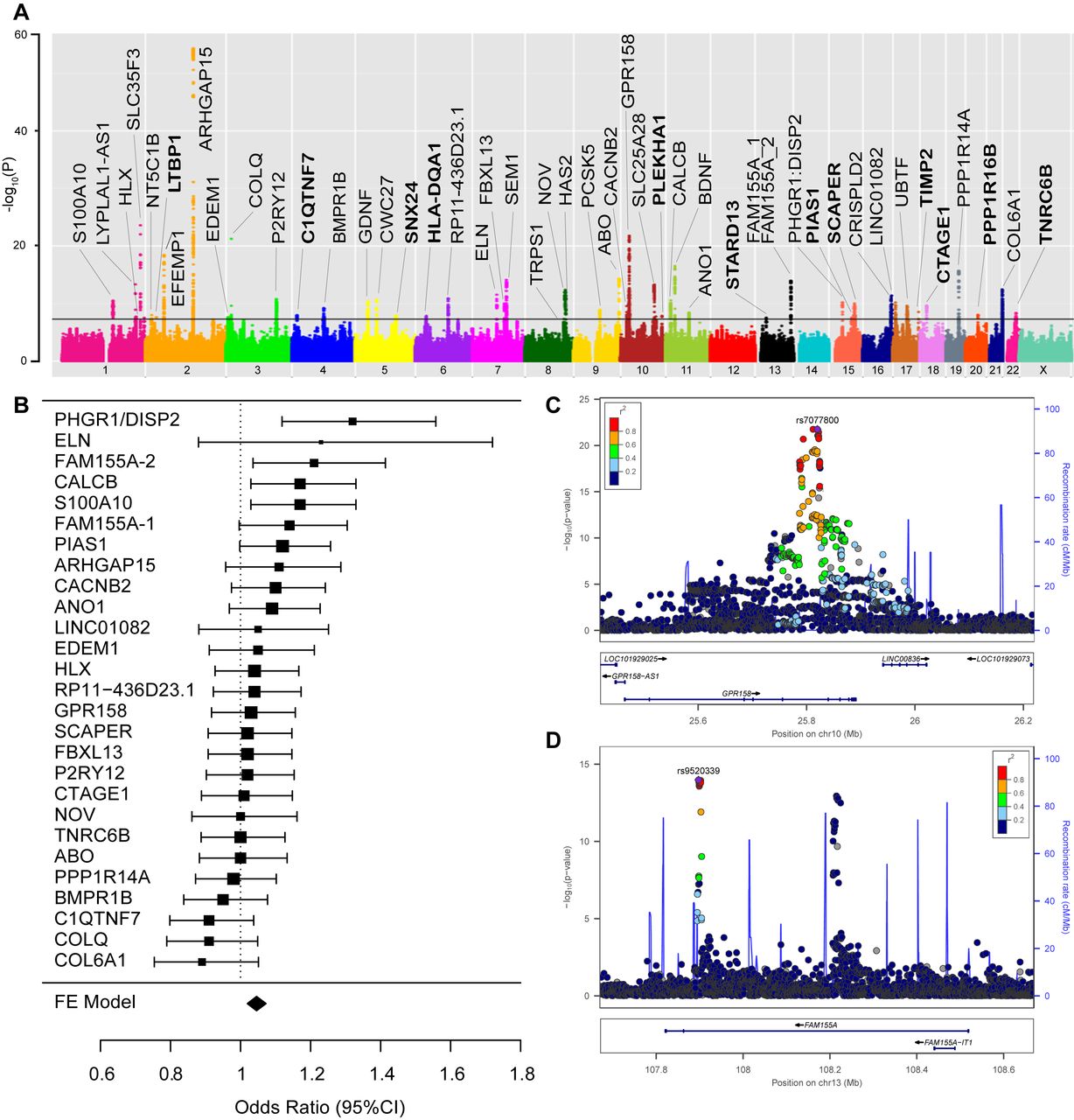
We observed genome-wide significant association (p<5×10−8) with diverticular disease for 2568 variants mapping to 51 independent genomic loci (online supplementary table 1), of which 12 had not been previously discovered (table 2). The resulting Manhattan plot is shown in figure 1A. The genomic inflation factor (λGC) was 1.199 and after LD score regression, the intercept was 1.02—an acceptable level for this size of study (QQ plot in online supplementary figure 1).23 The 51 loci were validated in a combined European sample of 3893 cases and 2829 diverticula-free controls based on colonoscopy (table 1). The direction of genotypic effect between discovery and replication samples was consistent for 48 out of 51 loci (93.8%; p for binominal test=1×10−9) (online supplementary table 5) and ORs were strongly correlated between both analyses (r=0.87; p=1.59×10−13, online supplementary figure 2). Nominal replication significance (p<0.05) and a consistent direction of effect between the two cohorts were observed for 27 loci within European colonoscopy cohorts (online supplementary table 6). Additional replication was observed for further eight loci in a combined meta-analysis of European colonoscopy cohorts with a European population cohort from Michigan (tables 2 and 3). Thirty-six out of 48 identified risk loci have been previously reported15 with genome-wide significant association (tables 2 and 3 and online supplementary table 4). All previously replicated risk loci for diverticular disease (ARHGAP15, FAM155A, COLQ) and (GPR158, ABO, ANO1/FADD, ELN, BMPR1B, SLC35F3, SEM1/SHFM1) were identified both in the current GWAS and replication analyses with similar ORs to those reported by Sigurdsson et al 14 and Maguire et al 15 (table 3). The most significant novel risk variant rs9960286 is located near CTAGE1 (cutaneous T-cell lymphoma-associated antigen 1) with a p value of 2.3×10−10 and 0.002 (ORallelic=1.14 (95% CI 1.05 to 1.24)) in the replication analysis. The most significant novel replicated risk variant rs60869342 is located in NOV (nephroblastoma overexpressed) with a p value of 4.4×10−13 and 0.0003 (ORallelic=0.85 (95% CI 0.78 to 0.93)) in the replication analysis; rs1381335 (r²=0.81 to rs60869342) in NOV was reported previously by Maguire et al 15 as risk locus #21, however, without formal replication.
Supplemental material
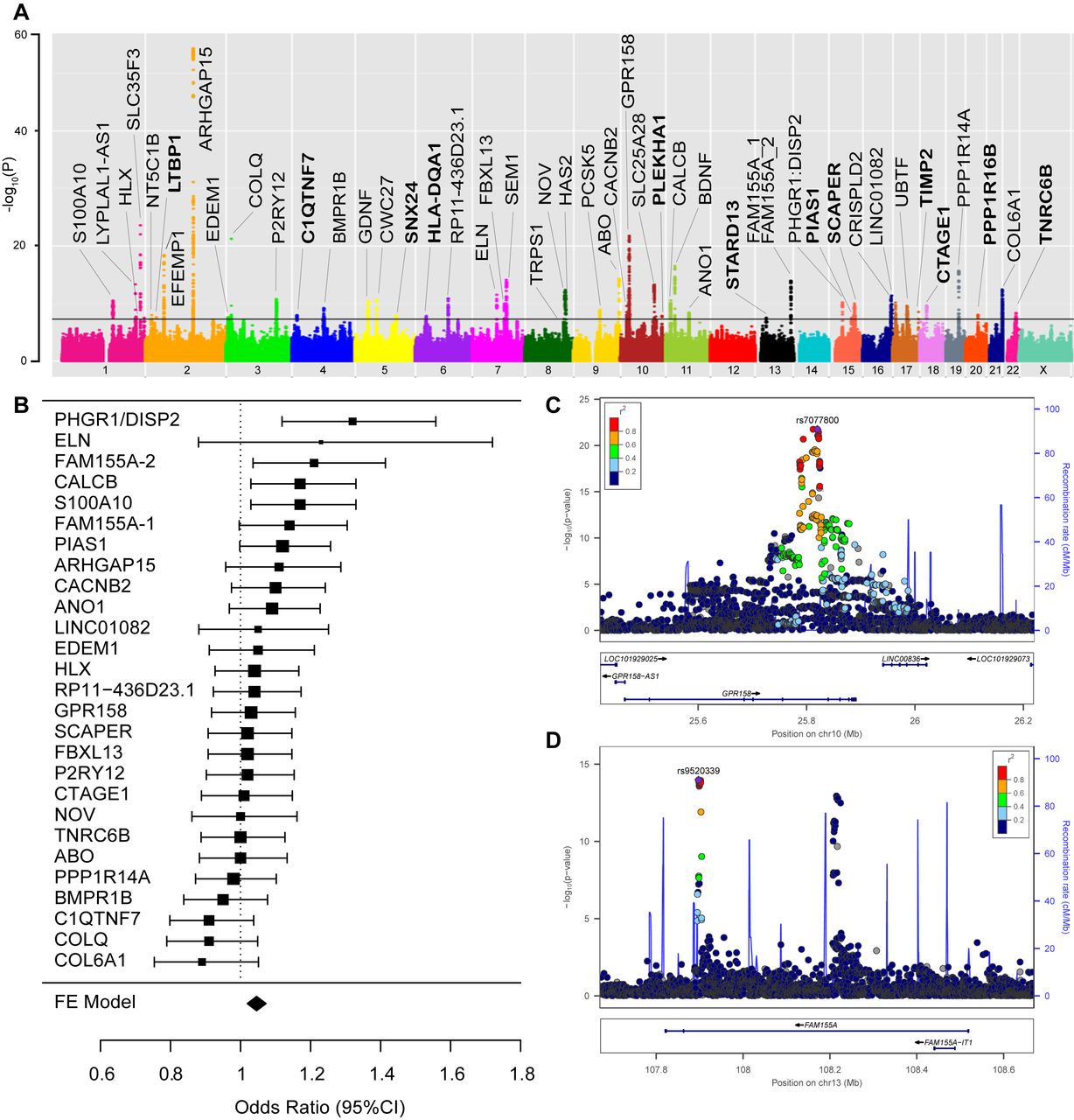
Genome-wide association study (GWAS) results. Principal findings of genetic analyses: panel A: Manhattan plot of genome-wide association results for diverticular disease. P values (−log10) are shown for SNPs that passed quality control. The genome-wide significance threshold (5×10−8) is shown as a black line. Gene names for loci with consistent effect and a known gene annotation are included in the panel. Gene names for newly discovered loci (as detailed in table 2) are printed in bold. Panel B: forest plot with 95% CIs of the relative impact of the 27 replicating variants on diverticulitis vs diverticulosis risk. ORs >1 indicate a higher impact on diverticulitis risk. The respective reference allele is provided in online supplementary table 17. Panel C: locus plot for diverticular disease risk locus GPR158. The −log10 (p values, mixed model association test) are plotted against SNP genomic position based on NCBI Build 37, with the names and location of nearest genes shown at the bottom. The variant with the lowest p value (lead variant) in the discovery analysis in the region is marked by a purple diamond. SNPs are coloured to reflect correlation with the most significant SNP, with red denoting the highest LD (r2>0.8) with the lead SNP. The association signal is confined to a single association peak located intronic in GPR158. Estimated recombination rates from the 1000 Genomes Project (hg19/genomes March 2012 release, EUR population) are plotted in blue to reflect the local LD structure. Gene annotations were obtained from the UCSC Genome Browser. The plot was generated using LocusZoom. Panel D: locus plot for diverticular disease risk locus FAM155A: the variant with the lowest p value in the FAM155A-1 region is marked by a purple diamond. For the FAM155A gene, two independent association signals (termed FAM155A-1 and FAM155A-2) with low pairwise LD (r2=0.0043) were considered as individual loci. SNPs are coloured to reflect correlation with the most significant SNP at FAM155A-1, with red denoting the highest LD (r2>0.8) and dark blue the lowest LD (r2<0.2) with the lead SNP.
Post hoc analysis of diverticulitis risk
The 27 replicating loci within European colonoscopy cohorts were evaluated for their relative genetic impact on diverticulitis (n=1167) and uncomplicated diverticulosis (n=1756) in a subset of the replication samples with the respective subphenotype information (online supplementary table 16). The majority of loci showed similar odds ratios for diverticulosis and diverticulitis (figure 1B, online supplementary table 17). Based on a 95% CI, four loci showed stronger effects for diverticulitis, namely variants at PHGR1 (OR 1.32, 95% CI 1.12 to 1.56), FAM155A-2 (OR 1.21, 95% CI 1.04 to 1.42), calcitonin-related polypeptide beta (CALCB) (OR 1.17, 95% CI 1.03 to 1.33) and the S100A10 (OR 1.17, 95% CI 1.03 to 1.33) locus.
Real-time PCR and immunohistochemistry analysis of curated candidate genes
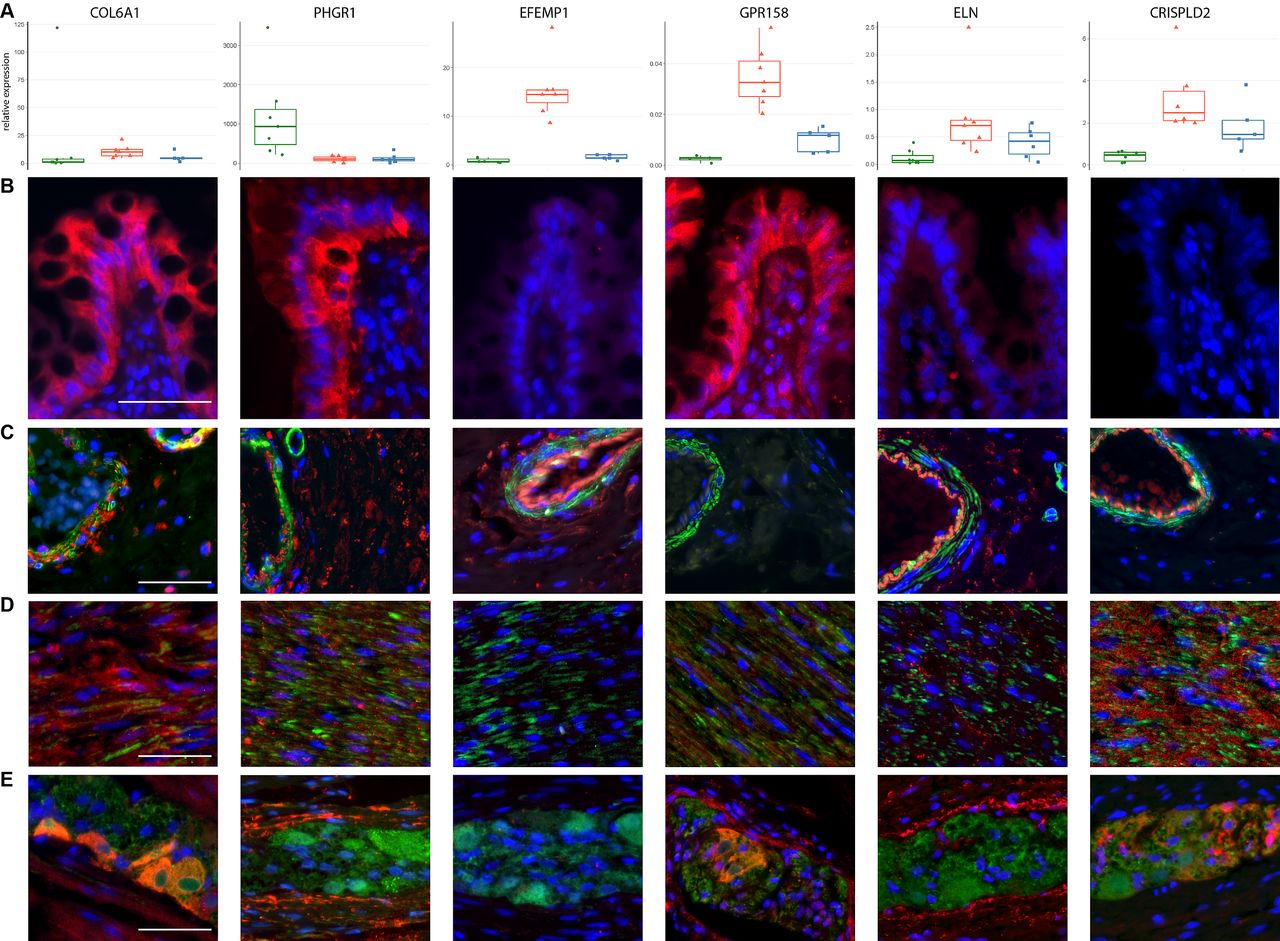
We next selected candidate genes for further experimental analysis as detailed for each locus in online supplementary table 3. Except for locus #25 (online supplementary table 1, PHGR1 and DISP2), a single curated candidate gene ‘lead candidate gene’ was assigned to each locus, based on local LD structure and supporting evidence from regulatory elements (eQTL and chromatin interaction). To provide a first indication of the relevant microanatomical colonic compartment relevant for disease, transcripts encoded at the top 20 replicating loci (online supplementary table 6) were analysed by quantitative real-time PCR in RNA preparations of the mucosal, submucosal and muscular layer from seven control patients (online supplementary table 7A,8). The majority of transcripts (13 out 18 at p<0.05) showed layer-specific expression patterns indicating the relevance of this higher histotopographical resolution as compared with total colonic expression (online supplementary table 9, supplementary figure 3). A potential disease-specific regulation of transcripts within each the mucosal, submucosal and muscular layer was analysed in 20 controls, 13 diverticulosis and 21 diverticulitis patients (online supplementary table 7b). A trend for upregulation of S100A10 (nominal p=0.003) in the submucosal layer in diverticulitis patients was noted, while overall a primary and strong disease-specific differential expression finding was not observed (online supplementary table 10 and supplementary figure 4). To obtain further spatial resolution, the localisation of expressed protein at selected novel loci with expression in all layers (COL6A1), predominant expression in the mucosa (PHGR1), submucosa (GPR158, EFEMP1) and submucosa and muscle layer (ELN, CRISPDL2) was investigated by immunohistochemistry (figure 2B-E). As epitomised for instance for GPR158, which localises predominantly to enteric ganglia and mucosa or elastin (ELN), which localises to the lamina propria, vessel walls and muscle, significant additional information is gained by this higher anatomical resolution.
{kind=link}
{kind=link}
Expression of candidate genes. Layer-specific expression pattern of novel candidate genes for diverticular disease. Panel A: normalised mRNA expression in the mucosal (left, green), submucosal (middle, red) and muscular (blue, right) layers in control colon (n=7). Panels B–E: fluorescence immunohistochemical analysis of expression in control colon in the mucosa (B), submucosal (C), muscular layer (D) and in myenteric ganglia (E). The respective target gene antibody is labelled in red, with DAPI (blue) for nuclear staining and alpha smooth muscle actin (smooth muscle marker, (C, D) and Protein Gene Product 9.5 (neuronal marker, (E) in green. It is evident that candidate genes show different expression patterns within the colonic wall and are localised to specific structures such as blood vessels, lamina propria, epithelium, smooth muscle or nerve cells. Scale bars are added in white (50 µm).
Overlap with IBD, IBS and monogenic syndromes
There was no overlap of the 2568 genome-wide significant variants (p<5×10−8) for diverticular disease with the 634 reported risk variants (p<9×10−6) according to the GWAS catalogue24 for IBD, Crohn’s disease (CD) and UC. Also, there was no overlap of the lead candidate genes at the 48 risk loci within the GWAS catalogue reported risk genes for IBD, CD and UC,25 except for HLA-DQA1. However, the IBD lead variant rs6927022 at the HLA-DQA1 locus was not in LD to the diverticular disease associated lead SNPs according to FUMA, thus pointing to a non-overlapping genetic risk structure. The percentage of individuals diagnosed with IBS among GWAS cases was 7.6% as compared with 3.1% among controls not diagnosed with diverticular disease. None of the 51 genome-wide significant lead variants for diverticular disease was significantly associated with the IBS phenotype in the UK Biobank (15 401 diagnoses of IBS vs 4 06 175 controls without a diagnosis of diverticular disease and without a diagnosis of IBS, data not shown). In contrast, mutations in 12 of the lead candidate genes for diverticular disease are reported in OMIM26 as autosomal dominant or recessive causative factors for 18 monogenic syndromes (online supplementary table 18). Many of these genes fall into the broad categories of neuromuscular syndromes, connective tissue stability disorders and morphogenesis traits and are considered in depth in the ’Discussion' section. A hypothesis-free analysis of the overlap of the genomic risk locations for diverticular disease within 500 kb distance to the lead variant is provided in online supplementary table 19.
Functional implications of curated candidate gene signature
Consistent with the overlap with monogenic syndromes, a gene set enrichment analysis (GSEA/MSigDB)21 using the 48 lead candidate genes revealed significant enrichments for neuromuscular mechanisms, connective tissue strength and morphogenesis (online supplementary table 11, online supplementary figure 5) and significant overlap with extracellular matrix-associated proteins of the murine colon (online supplementary table 12).
Functional implications based on in silico analysis of the global diverticulosis risk signature
We performed additional hypothesis-free functional and gene annotations based on the genomic positions of risk loci using FUMA17 as the curated candidate genes might not capture the full biology of the risk architecture. Positional gene mapping aligned SNPs to 176 genes, eQTL gene mapping matched cis-eQTL SNPs to 269 genes whose expression levels they influence (snp-gene pairs with FDR<0.05), with 21 genes specifically affected in sigmoid colon (online supplementary table 20). Chromatin interaction mapping annotated SNPs to 977 genes based on three-dimensional DNA-DNA interactions. This resulted in 1080 unique mapped genes (online supplementary table 2 and online supplementary figure 6). The majority of these mapped genes were protein coding genes (61%), while 39% were RNA and pseudogenes. A graphical representation of all mapped genes is given as circular plots for each chromosome carrying a risk locus in online supplementary figure 7.
Using a broad definition of candidate variants, namely a p value cut-off of 1.0×10-5 and r2≥0.6 to an independent significant SNP at the diverticular disease risk locus, most variants were located either intronic or intergenic (online supplementary table 1). Eighteen variants, of which nine were genome-wide significant, constituted exonic non-synonymous variants (online supplementary figure 8, supplementary table 17). Based on the Combined Annotation-Dependent Depletion (CADD) score, the most likely variants with functional consequences were rs1042917 (COL6A2) and rs17855988 (ELN) with CADD scores of 25.8 and 23.2, respectively (online supplementary table 21). Detailed fine-mapping plots of each risk locus are provided in online supplementary figure 9 showing local LD structure to the lead variant and annotation of variants by potential pathogenic and functional consequence assessed by CADD score and Regulome score and presences of cis-eQTL variants in sigmoid colon tissue. At genomic risk locus #15 (table 2), our annotated candidate gene was COL6A1 with the lead SNP located intronic to the gene, instead of COL6A2 as implicated by the functional effect of the candidate SNP rs1042917. The proteins synthesised by both genes are subunits of collagen VI, thereby pointing to a consistent functional mechanism. The identification of the mechanistically causal variants at each risk locus will, however, require further experimentation in model organisms and human tissue.
Interestingly, 94.6% (4738 of 5007 SNPs) of candidate SNPs were located at sites of open chromatin (online supplementary figure 10). Because the majority of lead variants were located in non-coding regions and thus not directly amendable to functional interpretation, we used GARFIELD to analyse enrichment statistics for the diverticular disease GWAS risk dataset with cell-specific coding, non-coding and functional elements from the GENCODE, ENCODE and Roadmap projects.17 A graphical summary of the enrichment of DNAse I hypersensitive sites is provided in online supplementary figure 11. As reported in detail in online supplementary table 15, regulatory elements from fibroblasts, fetal muscle and brain were particularly enriched in the genetic risk structure of diverticular disease. To further mine the genomic locations for functional implications, we performed a VEGAS2Pathway analysis,22 which pointed to processes involved in cell and organ differentiation and extracellular matrix among the top five identified pathways (online supplementary table 13, 14).
Discussion
In this study, we report the largest and most detailed genome-wide analysis to date for diverticular disease. We discovered 48 risk loci with genome-wide significance and consistent OR in a replication sample. Twenty-seven of these loci replicate at a nominal significance level of p<0.05. Among these loci, 12 are novel risk loci for diverticular disease and 5 of the novel loci were also replicated in a European clinical cohort with detailed phenotyping and colonoscopy data for all controls. The three previously known risk loci14 ARHGAP15, COLQ and FAM155A are among the validated loci and support the robustness of the phenotype and analysis on both the previous study and our analysis. A recent study by Maguire et al 15, who analysed a smaller UK Biobank dataset (n=409 728 individuals) compared with the current study (n=451 099) identified 40 loci with genome-wide significance using GWAS results publicly available from the Roslin Gene Atlas. There was an overlap for 36 out of 48 identified loci with genome-wide significance between the studies. Maguire et al were able to replicate eight loci in an independent European population cohort from Michigan. We replicated further eight risk loci in a meta-analysis approach integrating data from this Michigan cohort. The current study thus increases the number of replicated susceptibility loci for diverticular disease to 35, of which 25 loci had previously not been replicated. A limitation of the discovery study is that controls were 4 years younger than the cases. The modest lower age of controls increases the chance to include yet undiagnosed cases in the control sample, thereby potentially reducing the statistical power of the GWAS analysis. We based the functional interpretation of the GWAS results both on curated candidate genes and on more inclusive automated analysis tools such as GARFIELD, VEGAS2 and FUMA. Both analysis strategies point to diverticular disease as foremost a disorder of intestinal neuromuscular function and impaired connective fibre support. Many of the risk genes implicated in polygenic diverticular disease also have been implicated in monogenic neuromuscular and connective tissue disorders, as will be detailed below, which was consistent with the pathway analyses. These findings provide a specific molecular basis for the previously suggested mechanisms of structural weakness of the intestinal wall and dysregulated intestinal motility. Additional risk loci point towards a relevance of intestinal epithelial and vascular function, while a prominent immune signature was not apparent in the data.
Neuromuscular mechanisms
A number of candidate genes point towards a dysfunction of the enteric nervous system and the neuromuscular junction in the large bowel. Mutations in COLQ cause myasthenic congenital syndrome and the gene product anchors asymmetric acetylcholine esterase in the basal lamina of the motoric endplate.27 COL6A1 encodes the alpha 1 subunit of collagen VI (ColVI).28 ColVI is required for the structural and functional integrity of the neuromuscular junction.29 Mutations in glial cell line-derived neurotrophic factor (GDNF) have been suggested to act in concert with RET mutations to produce aganglionic megacolon (Hirschsprung’s disease), which is characterised by congenital absence of intrinsic ganglion cells in the myenteric and submucosal plexuses of the GI tract.30 Impaired GDNF function has been shown at gene and protein level to occur in diverticular disease and during early stages of diverticula formation.31 Plausible links to neuronal physiology are also evident for GPR158, a G-protein coupled orphan receptor32 and brain-derived neurotropic factor.
Three identified genes point to calcium sensitisation and calcium-dependent signalling in GI smooth muscle33: inhibiting myosin light chain phosphatase activity with protein kinase C-potentiated phosphatase inhibitor protein-17 kDa (CPI-17, PPP1R14A) is considered one of the primary mechanisms underlying myofilament Ca2+ sensitisation.34 Furthermore, for ANO1 (anoctamin 1), a calcium-activated chloride channel, a role in mediating cholinergic neurotransmission in the murine gastric fundus has been shown.35 CACNB2 (Cav1.2) encodes for the beta-2 subunit of a calcium-dependent calcium channel. The expression of Cav1.2 channels in colonic smooth muscle cells is key to colonic motility, decreased in colonic inflammation and a potential treatment target for motility disorders.36 Taken together, these data give further evidence for disturbed enteric neuromuscular functions as a relevant mechanism of diverticular disease.2 37
Neuromuscular development
HLX is a homeobox transcription factor gene conserved across species.26 Mutations in HLX have been observed in two fetuses with congenital diaphragmatic hernia and HLX homozygous null mice have a short bowel and reduced muscle cells in the diaphragm.38 39 HLX homozygous null animals exhibiting abnormal developmental of the enteric nervous system.38
Connective tissue function and morphogenesis
A second common functional theme of the identified risk loci is connective fibre function based in pathway, molecular function and syndrome associations. For instance, ELN encodes a protein that is one of the two components of elastic fibres which confer elasticity to organs and tissues. Mutations in ELN cause autosomal dominant cutis laxa.40 Mutations in bone morphogenetic protein receptor type 1B (BMPR1B) underlie autosomal recessive Hunter-Thompson41 type of acromesomelic dysplasia. EGF-containing fibulin extracellular matrix protein 1 (EFEMP1) has been associated with polygenic susceptibility to inguinal hernia42 and varicose veins.43 EFEMP1 encodes fibulin-3, an extracellular matrix protein. Efemp1(-/-) mice developed multiple large hernias including inguinal hernias. Histological analysis of Efemp1(-/-) mice revealed a marked reduction of elastic fibres in fascia.44 The fibulin family of protein has been associated with further connective tissue disorders. Mutations in fibulin-5 have been identified in patients with cutis laxa and mutations in fibrillin 1 cause Marfan syndrome. Interestingly, the N-terminal region of fibrillin-1 mediates a bipartite interaction with LTBP1.45 Variants in cysteine-rich secretory protein LCCL domain containing 2 (CRISPLD2) have been associated with non-syndromic orofacial cleft.46 47 A further example without association to genetic syndromes includes tissue inhibitor of metalloproteinases 2 (TIMP2), a peptidase involved in degradation of the extracellular matrix. The S100A10 protein regulates the remodelling of the extracellular matrix through plasmin-dependent activation of matrix metallopeptidase 9 (MMP-9) and plasminogen-dependent macrophage tissue invasion.48 49
Mesenteric vascular function
Diverticula occur predominantly at sites of preformed weakness in the intestinal wall, namely at sites of vascular entry through the muscle layer. In the interaction between muscular layer and the vessel, vascular biology and contractility may play an additional role. CALCB, which plays a role in mesenteric vascular smooth muscle function50 and protein phosphatase 1 regulatory subunit 16B (PPP1R16B), which regulates endothelial cell function51 may provide a potential mechanistic basis for altered vascular biology at these entry points.
Epithelial function and risk of diverticulitis
Interestingly, only one of the identified candidate genes—namely PHGR1—has a clear and exclusive link to epithelial function. Proline-rich, histidine-rich and glycine-rich protein 1 mRNA and protein are found to be expressed specifically in epithelial cells of intestinal mucosa as shown previously52 and in our immunohistochemistry analyses in figure 2 with the highest expression in the most mature and differentiated cells. PHGR1 showed the strongest effect size (OR 1.3 in comparison to uncomplicated diverticulosis) among the few loci associated with a higher risk of diverticulitis suggesting that for this complication of diverticular disease, indeed epithelial cell function may play a key role.
In summary, the novel genetic risk signature indicates that diverticular disease is a disorder of impaired intestinal neuromuscular function, impaired mesenteric vascular smooth muscle function and of impaired connective fibre support. We observe an intriguing convergence of previous monogenic findings with the polygenic risk signature of diverticular disease through the overlap with syndromic neuromuscular, connective tissue and morphogenesis disorders. Through the phenotype and the established cell biology of the Mendelian syndromes, inference of the functional implication of the novel risk loci, for instance, at the motoric end plate is possible. The manifestation of the inflammatory complication—diverticulitis—in turn may be triggered by epithelial dysfunction in the context of altered colon anatomy. These findings provide a deeper understanding of colonic biology and disease pathophysiology and open a new path for a functional dissection and therapeutic tackling of this common disease.
Acknowledgments
The authors would like to thank all study participants, researchers, clinicians and administrative staff who contributed to this study.
References
Footnotes
CS, JWH and SB contributed equally.
TW, CD, AG, MNW and JH contributed equally.
Contributors JWH, SB, CL, FC: performed the experiments, analysed the data and wrote the manuscript; JWH, AH, SB, AR, WR, NB: performed the bioinformatic analyses; CL, FC, MB: performed real-time PCR, histological, immunohistochemical analyses; CS, FL, LK, MZ, WvS, MCR, JR, TB coordinated, managed collection of samples, performed phenotyping; WL coordinated and supervised collection of samples; SN, UH-S, FR, PH, BS, WK, JT, MZ, JR, AW-B, TJ, JK, MS, IV, PS, HB, HH, AV, J-UE, GB, AH, S H, SW, ML, TK, SB, UH-S, LP, LSN, H-WS, SZ, SP, GF, AA, PTS, GL, JW, FL, TB, LK, PM, RG, VM: obtained the samples, performed phenotyping, interpretation of data; MK, MD’A, SZ, AF, MB, HV, WK, FL, RVT, JT gave conceptual advice, participated in the discussions, interpretation of the results, editing of the manuscript; CS, JH, MW, CD, AG, TW: conceived the experimental and analytical design, analysed data, wrote and reviewed the manuscript. All authors critically revised and contributed to the final manuscript.
Funding The work presented in this manuscript was supported by the German Research Council (DFG, Ha3091/9-1, WE2366/5-1) and the Austrian Science Fund (FWF, I1542-B13). Further support was received from SPAR Austria and from institutional funds from the Christian-Albrechts-University Kiel. The recruitment of the West German cohort was supported by a grant from the Faculty of Medicine, Saarland University (HOMFOR grant T201000747) to MCR. This study was supported by a grant of the Research Council of Lithuania No. SEN-06/2015/PRM15-135. AA, PTS were supported by the Stockholm County Council (ALF project). MD’A was supported by the Swedish Research Council (VR grant 2017-02403). Data access to the UK Biobank data was granted under project numbers 22691 and 9055.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.