Article Text

Abstract
Pancreatic ductal adenocarcinoma (PDA) is an almost uniformly lethal disease. One explanation for the devastating prognosis is the failure of many chemotherapies, including the current standard of care therapy gemcitabine. Although our knowledge of the molecular events underlying multistep carcinogenesis in PDA has steadily increased, translation into more effective therapeutic approaches has been inefficient over the last several decades. Evidence for this innate resistance to systemic therapies was recently provided in an accurate mouse model of PDA by the demonstration that chemotherapies are poorly delivered to PDA tissues because of a deficient vasculature. This vascular deficiency correlated with the presence of a dense stromal matrix that is a prominent histological hallmark of PDA tumours. Therapeutic targeting of stromal cells decreased the stroma from pancreatic tumours, resulting in increased intratumoral perfusion and therapeutic delivery of gemcitabine. Stromal cells contained within the PDA tumour microenvironment therefore represent an additional constituent to neoplastic cells that should be critically evaluated for optimal therapeutic development in preclinical models and early clinical trials.
- Pancreatic cancer
- tumour microenvironment
- tumour-stroma interactions
- genetically engineered mouse models of pancreatic cancer
Statistics from Altmetric.com
- Pancreatic cancer
- tumour microenvironment
- tumour-stroma interactions
- genetically engineered mouse models of pancreatic cancer
Introduction
Pancreatic ductal adenocarcinoma (PDA) is one of the most lethal human malignancies.1 The rapid clinical decline commonly observed in patients with pancreatic cancer has been ascribed to both the aggressive biological nature of PDA and to the ineffectiveness of systemic therapies available for patients with advanced disease.2 3 Indeed, even among patients with pancreatic cancer who undergo surgery and adjuvant chemotherapy, the median survival rate for those with clean microscopic surgical margins (R0 resection) is approximately 2 years, with a 5-year survival of 15–20%.4–6 One explanation for the poor response of patients to systemic therapies was recently provided in an accurate mouse model of PDA by the demonstration that chemotherapies are poorly delivered to PDA tissues because of a deficient vasculature.7 This vascular deficiency correlated with the presence of the dense stromal matrix that makes up the bulk mass of PDA tumours, and chemical inhibition of stromal cells decreased the matrix and increased intratumoral perfusion and therapeutic delivery. Stromal cells contained within the PDA tumour microenvironment therefore represent an additional constituent to neoplastic cells that should be critically evaluated for optimal therapeutic development.
Pancreatic carcinogenesis is currently understood as a multistage process characterised by the accumulation of genetic alterations accompanied by typical morphological and histological changes in pancreatic ductal cells. Activating mutations in the K-ras gene occur early during malignant transformation, followed by subsequent somatic mutations involving the tumour suppressor genes p16, p53 and DPC4.8 9 In addition, approximately 10% of all patients have an inherited predisposition to the development of PDA, and this has been partially ascribed to several germline mutations including BRCA2, STK11/LKB1, p16/CDKN2A and PRSS1.10 On the basis of these histopathological and molecular studies, a model similar to that of the adenoma-carcinoma sequence in the development of colon cancer11 was proposed to describe the progression from normal pancreas via preneoplastic lesions to invasive cancer.9 According to their stepwise accumulation of histopathological and molecular alterations, the preneoplastic lesions have been classified as pancreatic intraepithelial neoplasms (PanINs) 1a/b, 2 and 3.
The knowledge of high-grade PanIN lesions that are known to be a risk for developing PDA has led to early pancreatectomy; however, the impact of survival on such patients is currently unknown,12 and the extension of this concept to the general population has not been feasible. In addition, there have yet to be any effective molecular therapies reported based upon our knowledge of the PanIN progression scheme.
After decades of intensive efforts in genomic research focusing on molecular alterations in tumour cells,13 attention has increasingly expanded to include the tumour microenvironment, in particular the stromal cells. Many epithelial tumours including breast, prostate and ovarian cancers exhibit a prominent desmoplastic reaction with accumulation of stromal cells. Among them, pancreatic cancer displays the most extensive stromal reaction accounting for up to 90% of the tumour volume. Several notable studies have provided evidence that the microenvironment co-evolves with transformed epithelial cells in different carcinomas.14–20 However, the pathophysiological mechanisms of tumour stromal signalling and its contribution to tumour progression and therapeutic resistance are still poorly understood in pancreatic cancer and other solid carcinomas.21–23 Here we discuss recent preclinical models that hold great promise as they allow new concepts for improving the efficacy of chemotherapeutics to be tested rapidly and with high fidelity.
Stromal microenvironment in PDA
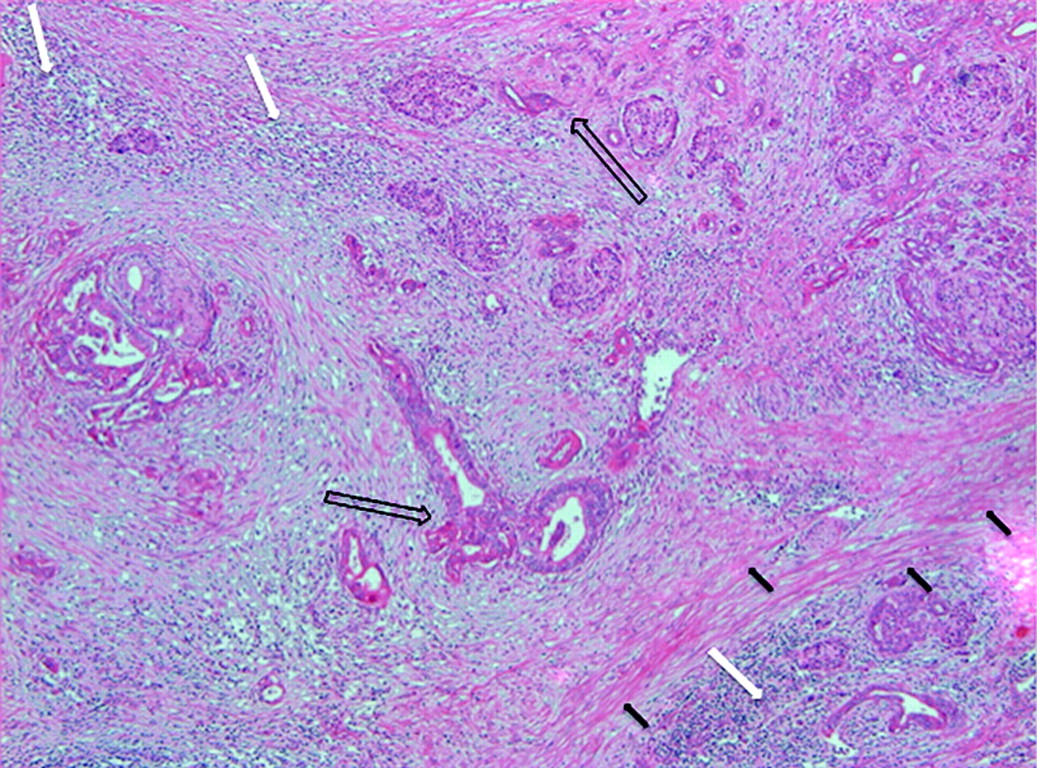
One of the most prominent histological features of PDA is the presence of an abundant tumour stroma (figure 1).24 The stromal microenvironment is a complex structure composed of an extracellular matrix (ECM), activated fibroblasts and myofibroblasts, inflammatory cells and blood and lymphatic vessels that distort the normal architecture of pancreatic tissue. Interactions between the neoplastic and non-neoplastic cells and acellular matrix have been proposed to stimulate the extensive desmoplastic reaction. At the molecular level, stroma production is promoted by the activation of multiple cancer cell-derived signalling pathways such as transforming growth factor β (TGFβ), hepatocyte growth factor (HGF/Met), fibroblast growth factors (FGFs), insulin-like growth factor 1 (IGF-1) and epidermal growth factor (EGF) via autocrine and paracrine mechanisms.25 26 These receptor-mediated signalling cascades lead to secretion of structural matrix components including proteoglycans, collagens and fibronectin as well as catalytically active enzymes such as proteinases.

H&E stain of human pancreatic ductal adenocarcinoma showing a prominent desmoplastic reaction (black arrows), neoplastic ductal cells (arrows) and inflammatory cells (white arrows).
The exact composition of the ECM is regulated by a multitude of different mechanisms. For instance, matrix metalloproteinases (MMPs) are a large family of zinc-containing proteolytic enzymes involved in the degradation, dynamic remodelling and turnover of ECM proteins in physiological and pathological conditions.27 In particular, MMP-2 and MMP-9 are commonly overexpressed in pancreatic cancer and play an important role in tumour cell migration and invasion by degrading the surrounding ECM.28 29 Recent evidence suggests that the interplay of extracellular proteinases and their inhibitors in invasion and metastasis is much more complex than previously anticipated. In particular, the biological functions of proteinase inhibitors extend far beyond their roles as inactivators of their target proteinases. For example, tissue inhibitors of metalloproteinases (TIMPs), in particular TIMP-1 and TIMP-2, are frequently overexpressed in pancreatic cancer and various other malignancies, along with their target proteinases.30 31 Another example is the serine protease inhibitor SERPINE2 (protease nexin I), which is overexpressed in various gastrointestinal malignancies and promotes ECM production and local invasion of pancreatic tumours in vivo.32 In addition, a multitude of proteins has evolved which modulate the composition of the ECM. Among them, the ECM metalloproteinase inducer (EMMPRIN) stimulates MMP-1 expression in fibroblasts and is frequently overexpressed in various solid tumours correlating with tumour size, stage and prognosis in primary breast and ovarian cancer.33 In pancreatic cancer, EMMPRIN is expressed on the cell surface and supernatant of EMMPRIN-positive pancreatic cancer cell lines such as MiaPaCa and Panc1 induces MMP-2 synthesis in cultured pancreatic stellate cells (PSCs).34
The complex interplay between tumour cells and stroma also leads to distinct changes in the transcriptional programme of the cellular components within the stroma, such as activated fibroblasts, stellate cells and inflammatory cells, which in turn promotes cancer cell motility, resistance to hypoxia and stromal neovascularisation. These effects in stromal cells include altered integrin expression patterns, increased expression levels of cyclo-oxygenase 2, vascular endothelial factor A (VEGF-A), collagen I and hypoxia-inducible factor-1α.35–40
Recently, activation of the developmental sonic hedgehog (SHH) pathway has been identified as another mediator that promotes stromal desmoplasia.20 41 Binding of SHH ligands to the patched1 receptor relieves repression of the 12-transmembrane domain protein Smoothened (SMO), resulting in activation of the Gli family of transcription factors. SHH is overexpressed in neoplastic cells of human pancreatic tumours42 while downstream signalling is confined to the stromal compartment, forming a paracrine signalling axis from neoplastic to stromal cells.43 44
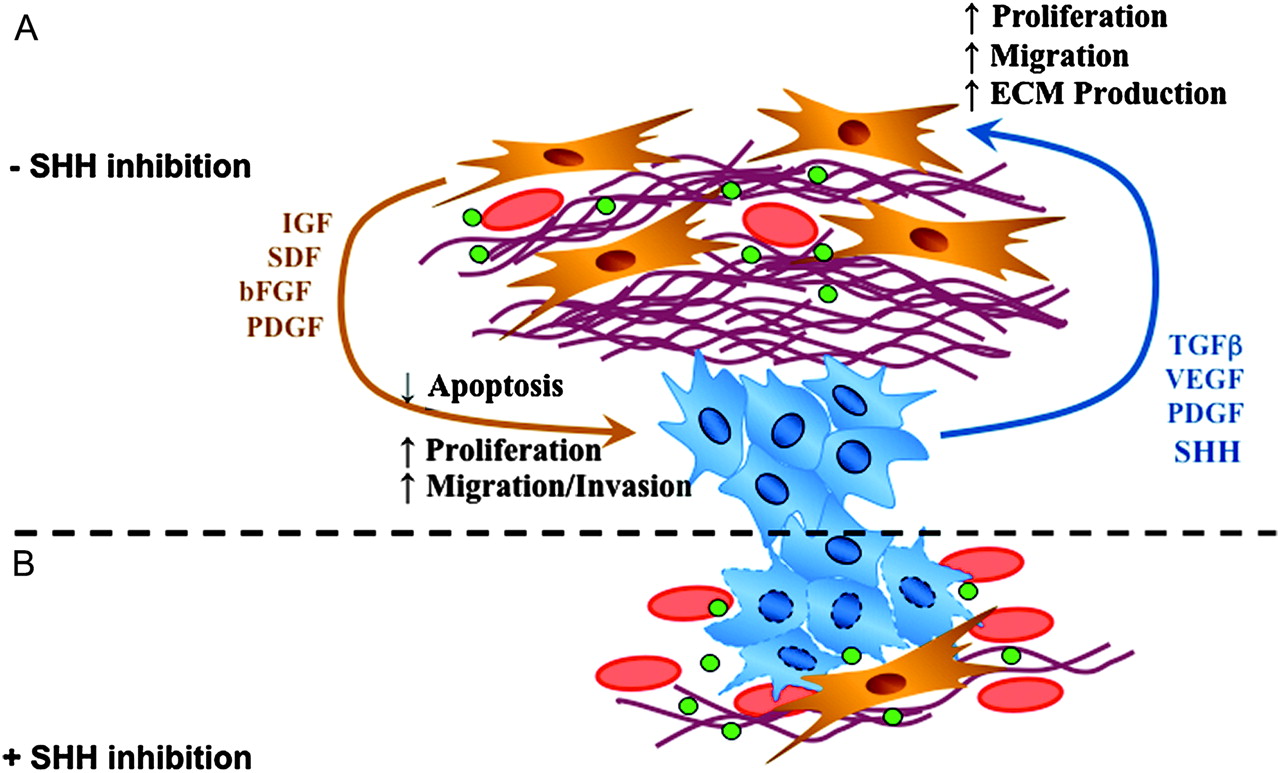
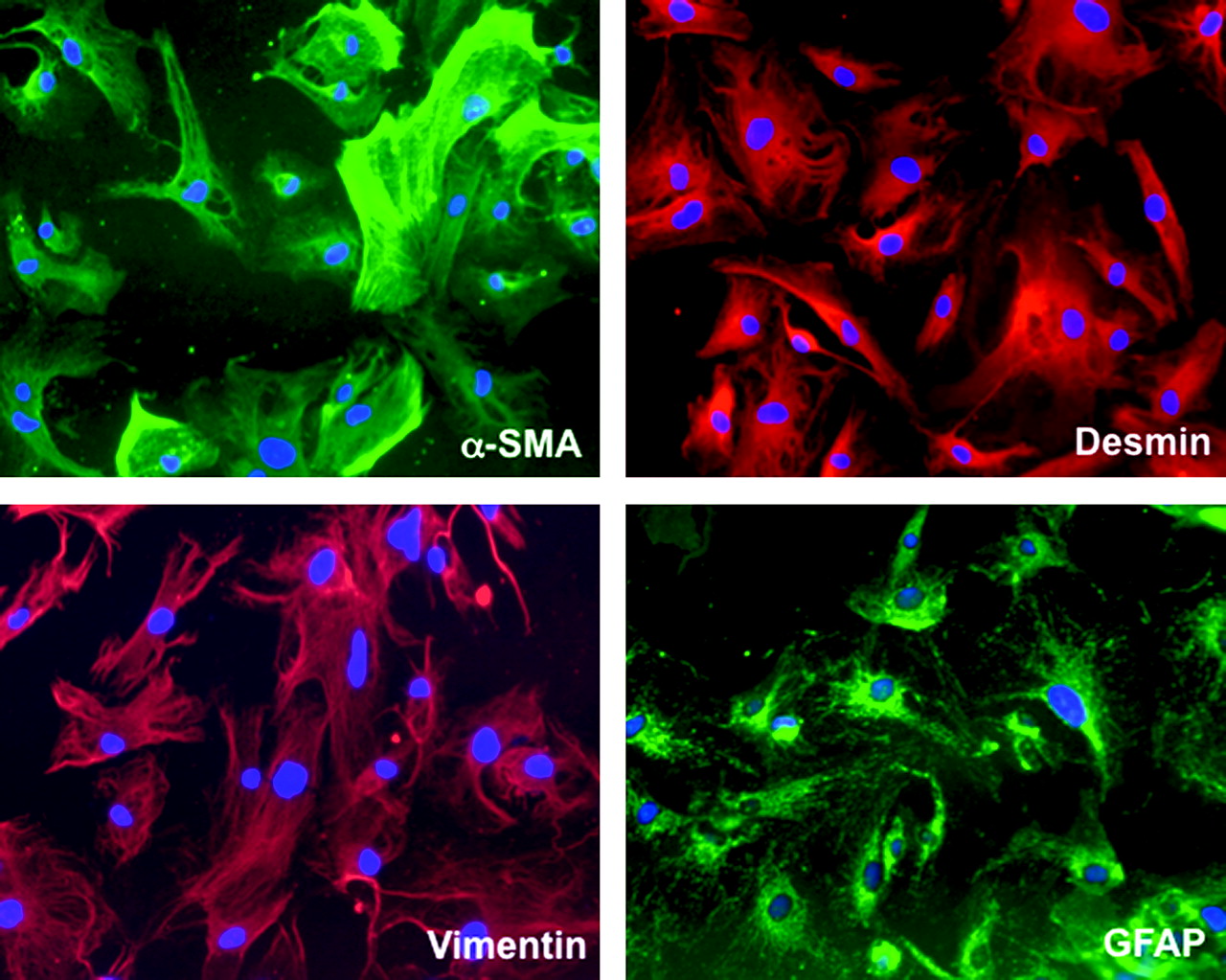
Interestingly, PSCs have emerged as pancreas-specific mesenchymal cells and important regulators of desmoplasia in pancreatic cancer.36 45 PSCs share many morphological and functional characteristics with hepatic stellate cells (HSCs) whose central role in liver fibrosis is well established. However, distinct differences in expression patterns were observed between HSCs and PSCs, reflecting organ-specific variations of the common stellate cell-specific phenotype.46 Isolation and in vitro culture of PSCs was first achieved in 1998 and provides a useful platform to investigate the mechanisms mediating epithelial–stromal interactions in pancreatic cancer.47 48 PSCs are normally located in the space between the acini and endothelial cells and store vitamin A as retinyl palmitate in lipid droplets.49 Two different functional stages can be clearly defined in PSCs—the quiescent state and the activated state (or ‘myofibroblastic’ state). In quiescence, PSCs store vitamin A droplets and are characterised by the presence of desmin and glial fibrillar acidic protein. Upon activation by growth factors, cytokines or oxidant stress, PSCs transform into a myofibroblast-like phenotype and secrete excessive amounts of collagen I, III, fibronectin and matrix degrading enzymes such as MMPs.48 Figure 2 shows immunofluorescence stains of cultured primary human PSCs. Notably, several studies suggest that activated PSCs rather than cancer cells are the main source of MMPs and TIMPs.50 51 Although proangiogenic molecules such as periostin and VEGF are secreted by PSCs, sustained PSC activation promotes fibrogenesis and ultimately may create a highly desmoplastic, hypovascular and hypoxic tumour microenvironment.52–54 Interestingly, pancreatic cancer cells induce PSC activation in vitro by growth factors such as TGF-β1, platelet-derived growth factor (PDGF) and VEGF.45 55 In vivo, co-injection of PSCs and cancer cells results in increased tumour growth accompanied by a pronounced desmoplastic reaction.55 56 A recent study using an orthotopic tumour model with co-cultured PSCs and pancreatic cancer cells showed increased migratory potential in both cell types but inhibition of apoptosis in cancer cells only, suggesting a pro-survival and pro-growth mutual interaction.57 Strikingly, stellate cells were also detected in metastatic foci in the liver of nude mice, suggesting co-migration of PSCs with cancer cells to establish a potentially tumour-favourable microenvironment at distant sites.57 Figure 3A schematically depicts the various critical pathways involved in the interaction between stromal cells and cancer cells.

Immunofluorescence stain of cultured primary human pancreatic stellate cells.
{kind=link}
{kind=link}
{kind=link}
(A) Schematic depiction of various pathways and growth factors (green dots) interacting between stromal cells (yellow) and cancer cells (blue). (B) Sonic hedgehog (SHH) inhibition decreases desmoplasia (fibrous reddish bundles) and increases the density of tumour vessels (red) around cancer cells (blue). ECM, extracellular matrix; FGF, fibroblast growth factor; IGF, insulin-like growth factor 1; PDGF, platelet-derived growth factor; SDF, stromal cell-derived factor; TGFβ, transforming growth factor β; VEGF, vascular endothelial growth factor.
The relevance of these findings is underscored by the histological evaluation of clinical specimens indicating that the prognosis and outcome of patients with pancreatic cancer heavily depends on the stromal activity and the ECM composition within the tumours. High activity of myofibroblasts as evidenced by immunohistochemistry against α-smooth muscle actin or secretion of distinct proteins such as secreted protein acidic and rich in cysteine (SPARC) were associated with a worse prognosis in patients with PDA, highlighting the impact of the stromal microenvironment on disease progression and patient survival.58–60
Taken together, the previously held notion that the tumour stroma of PDA is a defensive reaction of the host protecting against invasive growth and formation of metastases has been abandoned. In contrast, a highly dynamic tumour microenvironment is now being proposed that promotes tumour growth and invasion, protects from apoptosis and potentially creates barriers to the delivery of therapeutic compounds.35 36 61 62
In vivo modelling of tumour microenvironment: genetically engineered mice in PDA
Various mouse models of pancreatic cancer have been developed in the past few years, providing a crucial platform for the investigation of basic biological principles of cancer development and tumour biology.63 More recently, mouse models of cancer have been increasingly employed to investigate and discover novel preclinical and clinical anticancer agents.
Historically, the most commonly used animal models for PDA were xenograft tumours in immunodeficient mice generated by subcutaneous injection or orthotopic transplantation of tumour cell lines. These models are relatively simple to establish and human cancer cells can be assessed in the murine in vivo environment. Recently, primary patient-derived tumour xenografts have been described as a platform to develop personalised drug screening.64–66 However, major drawbacks of xenograft models include the impaired immune response owing to the need to use immunocompromised mice as hosts, the inability to perpetuate the human tumour microenvironment and the profound differences in tumour structure and vasculature compared with endogenous human PDAs.67 Accordingly, results obtained from a number of xenograft studies have not translated well into the clinic. For instance, PDA xenografts often respond well to anti-angiogenic agents,68 but these same agents often fail to show any clinical benefit in the cognate human tumour.69
An important milestone in PDA research was therefore the development of genetically engineered mouse models (GEMM).63 Of special interest are mutant mice that have been engineered to lose the expression of tumour suppressor genes (TSGs) or express oncogenes or dominant negative TSGs from their native promoters by using knock-out or knock-in technologies. To control and direct the spatiotemporal expression of the mutant alleles, site-specific recombinases such as Cre are used. Two GEMMs were recently developed that bear striking resemblance to human PDA. The first is based on mutation of the endogenous murine Kras gene specifically in pancreatic progenitor cells by crossing mice with a conditionally activated Kras allele (LSL-KrasG12D) to transgenic strains that express Cre recombinase in pancreatic lineages (PdxCre or p48Cre). These ‘KC’ mice develop murine PanIN lesions with 100% penetrance, but only a small subset of these animals progress to PDA at an advanced age, suggesting that additional genetic alterations are necessary for tumour formation.70 To accelerate the process of tumorigenesis, PdxCre-expressing compound mutant mice were generated with conditional mutations in both Kras and Trp53 in analogy to the genetic alterations in human PDA. These ‘KPC’ mice develop advanced PDA with 100% penetrance at an early age, thus recapitulating human PDA including histopathological similarities in neoplastic cells, desmoplasia, occurrence and site of metastasis and comorbidities such as cachexia, activation of biochemical pathways and evidence for genomic instability.71 Further important work was done by the Barbacid group providing striking evidence that temporal expression of endogenous Kras in acinar cells of adult mice results in PanINs and invasive PDA only in the context of caerulein-induced pancreatitis.72 Thus, GEMMs (in contrast to xenograft models) are particularly suited to elucidate the role of the tumour-microenvironment interactions in the disease initiation and progression of pancreatic cancer. Furthermore, preclinical studies can be established that examine the effects of drugs on the tumour microenvironment and specifically target tumour-associated stromal cells.
Stroma-targeted therapies in pancreatic cancer
Over the last decade, major efforts have been undertaken to enhance the effect of the current standard of care chemotherapy, gemcitabine, by the combination with a second cytotoxic drug.73 To this end, large randomised phase III trials were performed to evaluate additional effects of cisplatin,74 75 oxaliplatin,76 77 5-FU,78 irinotecan,79 80 exatecan81 and pemetrexed,82 but there was no significant overall survival benefit. For the combination capecitabine and gemcitabine versus gemcitabine alone, initial data suggest no significant advantage for overall survival.83 Notably, a recent phase III trial revealed a trend towards improved overall survival, and a meta-analysis involving 935 patients showed a significant survival benefit for the combination of capecitabine and gemcitabine.84 Until recently, the mechanism for the extremely poor responsiveness to therapeutic agents has been mainly ascribed to the heterogeneity of transformed cells rather than to the tumour microenvironment.
The improved knowledge of the genetic and molecular alterations not only occurring in tumour cells but also in the surrounding stromal cells has recently led to the development of novel therapeutic approaches specifically targeting profibrotic pathways, cytokines and growth factors involved in tumour desmoplasia and angiogenesis to control tumour growth, prevent formation of metastases and increase the cytotoxic effect of chemotherapeutics.
Following promising results from preclinical studies, marimastat, a broad-spectrum synthetic MMP inhibitor, was the first compound tested in a large randomised phase III trial in 414 patients with advanced pancreatic cancer. Initially fuelled with great enthusiasm, the results were rather disappointing as neither marimastat alone nor the combination of marimastat and gemcitabine showed any improvement in overall survival or tumour control compared with gemcitabine alone.85 86 One year later in 2003, Moore et al reported the results of a phase III trial with BAY-12-9566, a specific inhibitor of MMP-2, MMP-3, MMP-9 and MMP-13. Patients with locally advanced or metastatic pancreatic cancer were treated with BAY-12-9566 or standard intravenous gemcitabine; however, the study was discontinued after completion of the second interim analysis showed that the new substance was significantly inferior to gemcitabine (median overall survival 3.74 months vs 6.59 months).87 As pointed out earlier, the role of proteases in cancer biology, in particular MMPs, is highly complex and the failure of broad-spectrum anti-MMP therapies might at least partly be explained by the fact that MMPs have pro-tumorigenic as well as tumour suppressive functions.
As cardinal mediators of tumour neoangiogenesis, overexpression of VEGF and its receptors (VEGFR-1, VEGFR-2 and VEGFR-3) has been associated with poor prognosis and increased metastatic potential in pancreatic cancer.88 Bevacizumab, a recombinant humanised anti-VEGF monoclonal antibody approved for the treatment of colon cancer, has also been investigated in PDA. Despite promising results from a previous phase II trial,69 the combination of bevacizumab and gemcitabine failed to significantly prolong survival in a large phase III trial with 602 patients with pancreatic cancer.89 Similar discouraging results were obtained with a clinical phase II trial in 103 patients with pancreatic cancer using gemcitabine and axitinib, an oral inhibitor of VEGFR-1, VEGFR-2 and VEGFR-3.90
The only targeted agent which demonstrated a statistically significant effect on overall survival is erlotinib, a small molecule inhibitor of the EGFR tyrosine kinase. EGFR is frequently overexpressed in pancreatic cancer and correlates with poor prognosis and disease progression.91 EGFR signalling has also been shown to impact pancreatic stromal reaction by activating PSCs.92 The combination of gemcitabine and erlotinib conferred a marginal but significant improvement in survival over gemcitabine alone in a large phase III randomised trial (median survival 6.24 months vs 5.91 months).93
Taken together, numerous clinical trials have failed to substantially improve the prognosis of patients with advanced pancreatic cancer during the last decade. The general resistance of human PDA to systemic therapies in vivo is unusual compared with other solid carcinomas, casting doubt on the transferability of preclinical results to the clinical situation in PDA.94 It can be assumed that the lack of survival benefit shown by conventional and targeted agents in patients with pancreatic cancer might at least partly evolve from the predominant desmoplastic stroma reaction and the pronounced hypovascularity.
Indeed, experimental evidence was provided very recently demonstrating that the hypovascular tumour stroma affects delivery of chemotherapeutics in a GEMM of PDA. We showed that the active intracellular metabolite of gemcitabine, 2′,2′-difluorodeoxycytidine triphosphate (dFdCTP), was detectable in transplanted xenograft tumours but undetectable in tumours of KPC mice which are characterised by a pronounced desmoplastic reaction highly resembling the human PDA phenotype. Subsequent inhibition of the SHH signalling pathway by IPI-926, a semisynthetic derivative of cyclopamine, resulted in a dramatic depletion of stromal components paralleled by an increase in intratumoral vascular density. Although stroma depletion alone had no immediate antitumour effect in this experimental setting, co-administration of gemcitabine and IPI-926 resulted in a significantly enhanced intratumoral concentration of dFdCTP, transient disease stabilisation and a statistically significant prolongation of survival.7 However, the pronounced stromal reaction ultimately returned in the KPC model, suggesting that the tumours can adapt to chronic SHH inhibition.7 The effects of SHH inhibition on the stromal and vascular architecture are schematically displayed in figure 3B.
This study therefore provides a proof of principle that disruption of the desmoplastic stroma facilitates the delivery and enhances the efficacy of gemcitabine in PDA. Poor perfusion and a deficient non-angiogenic vasculature limits drug delivery and may also help to explain the recent failures of anti-VEGF strategies in pancreatic cancer.
Three months after we published these murine data, exciting clinical results on non-invasive quantification of blood flow and metabolic activity of pancreatic tumours using oxygen-15-labelled water [15O]-H2O and [18F]-fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT imaging were presented by a Finnish group.95 In this small study, pancreatic tumours were characterised by reduced blood flow and high metabolic activity compared with normal pancreatic tissue.95 Furthermore, a high ratio of glucose uptake to blood flow was a predictor of poor prognosis, further supporting the novel concept that a highly dynamic but hypovascular tumour microenvironment contributes to chemoresistance and poor therapeutic outcome in patients with pancreatic cancer by creating barriers for drug delivery.95
Conclusions
The desmoplastic hypovascular tumour microenvironment consisting of large amounts of ECM proteins, activated fibroblasts, stellate cells and inflammatory cells is now recognised to represent the cardinal histological hallmark feature of PDA. Recent preclinical and clinical data suggest that this stromal microenvironment creates a ‘fortress-like’ hypovascular barrier that impairs the delivery of chemotherapeutics and promotes aggressive neoplastic cell behaviour. The extremely poor prognosis and resistance to systemic therapies might therefore be partly explained by inefficient drug delivery to the tumour cells rather than drug resistance of the tumour cells. Breaching this ‘stroma fortress’ represents a promising strategy to improve the delivery and efficacy of systemic chemotherapeutics and might open new therapeutic avenues for patients with PDA. One challenge in translating these findings to the clinical care of patients is the need to develop means to accurately measure drug levels in PDA tumours with non-invasive techniques or small biopsies. However, clinical trial design is often hampered by the fact that PDA most commonly occurs in elderly people and is associated with severe cachexia and other age-related conditions. GEMMs of PDA are particularly suited to study the biology and treatment of this disease and may also be useful for developing novel pharmacokinetic approaches. GEMMs should also help define the role of PSCs in stimulating the desmoplastic stroma, and determine whether these are the target cells of hedgehog inhibitors. Additional pathways known to be involved in the activation of PSCs such as FGF, PDGF or IGF-1 should also be interrogated as potential therapeutic targets in the GEMM.61 96 Furthermore, efforts aimed at inducing PSC transdifferentiation from an activated to a quiescent state via administration of vitamin A analogues could also be an attractive modality as reported in culture-activated rat PSCs.96
However, as observed in the mouse PDA model, tumours adapt to chronic inhibition of profibrotic signalling and ultimately resume stromal desmoplasia and hypovascularity. We therefore anticipate that multiple approaches that target the PDA stroma will probably be necessary in order to circumvent this adaptive response and maximise therapeutic benefits for patients.
Key points 1: Basic science
A pronounced desmoplastic and hypovascular microenvironment is a hallmark feature in pancreatic cancer.
The stromal microevironment contributes to disease progression, promoting tumour growth and invasion.
Pancreatic stellate cells have emerged as pancreas-specific mesenchymal cells and key regulators of desmoplasia in pancreatic cancer.
The cellular and molecular mechanisms underlying the regulation and perpetuation of desmoplasia in pancreatic cancer are still poorly understood.
Key points 2: Clinical science
The devastating prognosis for patients with pancreatic cancer has remained virtually unchanged over the last three decades.
Targeting the stromal microenvironment is a novel and promising concept for tailored therapies in pancreatic cancer.
Genetically engineered mice are particularly suited to study the tumour microenvironment and test novel therapeutic regimens targeting the tumour stroma (eg, sonic hedgehog inhibition).
The extremely poor prognosis and the resistance to systemic chemotherapies are at least partly explained by low drug delivery caused by stromal barriers rather than epithelial tumour cell resistance itself.
The development of effective means to measure intratumoral levels of chemotherapy (eg, dFdCTP) with either non-invasive imaging or using reliable microscopic biopsies is an important challenge for the field of pancreatic cancer.
References
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.