Article Text

Abstract
Objective Monocyte chemoattractant protein-1 (MCP-1, CCL2), the primary ligand for chemokine receptor C–C chemokine receptor 2 (CCR2), is increased in livers of patients with non-alcoholic steatohepatitis (NASH) and murine models of steatohepatitis and fibrosis. It was recently shown that monocyte/macrophage infiltration into the liver upon injury is critically regulated by the CCL2/CCR2 axis and is functionally important for perpetuating hepatic inflammation and fibrogenesis. The structured L-enantiomeric RNA oligonucleotide mNOX-E36 (a so-called Spiegelmer) potently binds and inhibits murine MCP-1. Pharmacological inhibition of MCP-1 with mNOX-E36 was investigated in two murine models of chronic liver diseases.
Methods Pharmacological inhibition of MCP-1 by thrice-weekly mNOX-E36 subcutaneously was tested in murine models of acute or chronic carbon tetrachloride (CCl4)- and methionine–choline-deficient (MCD) diet-induced chronic hepatic injury in vivo.
Results Antagonising MCP-1 by mNOX-E36 efficiently inhibited murine monocyte chemotaxis in vitro as well as migration of Gr1+ (Ly6C+) blood monocytes into the liver upon acute toxic injury in vivo. In murine models of CCl4- and MCD diet-induced hepatic injury, the infiltration of macrophages into the liver was significantly decreased in anti-MCP-1-treated mice as found by fluorescence-activated cell sorting (FACS) analysis and immunohistochemistry. In line with lower levels of intrahepatic macrophages, proinflammatory cytokines (tumour necrosis factor α, interferon γ and interleukin 6) were significantly reduced in liver tissue. Overall fibrosis progression over 6 (CCl4) or 8 weeks (MCD diet) was not significantly altered by anti-MCP-1 treatment. However, upon MCD diet challenge a lower level of fatty liver degeneration (histology score, Oil red O staining, hepatic triglyceride content, lipogenesis genes) was detected in mNOX-E36-treated animals. mNOX-E36 also ameliorated hepatic steatosis upon therapeutic administration.
Conclusions These results demonstrate the successful pharmacological inhibition of hepatic monocyte/macrophage infiltration by blocking MCP-1 during chronic liver damage in two in vivo models. The associated ameliorated steatosis development suggests that inhibition of MCP-1 is an interesting novel approach for pharmacological treatment in liver inflammation and steatohepatitis.
- Monocyte
- Kupffer cell
- chemokine
- monocyte chemoattractant protein-1
- liver cirrhosis
- NASH
- liver immunology
- cancer genetics
- cell death
- chronic liver disease
- liver
- hepatocyte
- macrophages
- IL-6
- hepatitis C
Statistics from Altmetric.com
- Monocyte
- Kupffer cell
- chemokine
- monocyte chemoattractant protein-1
- liver cirrhosis
- NASH
- liver immunology
- cancer genetics
- cell death
- chronic liver disease
- liver
- hepatocyte
- macrophages
- IL-6
- hepatitis C
Significance of this study
What is already known about this subject?
Infiltration of monocytes/macrophages is a critical contributing factor for chronic liver inflammation and fibrosis in mice and men.
Monocyte/macrophage migration into the liver is primarily regulated by the chemokine monocyte chemoattractant protein-1 (MCP-1) (CCL2) and its corresponding receptor C–C chemokine receptor 2 (CCR2).
The MCP-1/CCR2 pathway is especially activated in patients with non-alcoholic steatohepatitis (NASH).
Inhibition of MCP-1 by a novel RNA oligonucleotide (Spiegelmer) is currently being investigated in patients with type II diabetes to ameliorate nephropathy.
What are the new findings?
Hepatic monocyte/macrophage infiltration can be specifically and efficiently blocked by mNOX-E36 (murine MCP-1-specific antagonist) in two independent models of chronic liver injury in mice.
Reduced hepatic macrophages upon MCP-1 inhibition result in lower intrahepatic levels of proinflammatory cytokines in injured liver.
Inhibition of MCP-1 significantly ameliorates steatosis progression, but does not affect hepatic fibrogenesis.
How might it impact on clinical practice in the foreseeable future?
Inhibition of MCP-1 efficiently reduces hepatic macrophage infiltration and is an interesting novel approach for pharmacological treatment in liver inflammation and steatohepatitis.
The MCP-1-specific ‘Spiegelmer’ compound is already in clinical trials with patients with diabetes type II and might have additional beneficial effects on hepatic co-morbidities, especially on steatosis and steatohepatitis.
Introduction
The consequence of chronic liver injury by various conditions such as metabolic disorders, viral hepatitis or alcohol abuse is a typical remodelling of the organ characterised by fibrosis and steatosis (ie, hepatocytic accumulation of fat).1 Hepatic steatosis is also commonly observed in systemic metabolic diseases such as diabetes, obesity and dyslipidemia. It often progresses to non-alcoholic steatohepatitis (NASH) with evidence of hepatocellular injury and chronic inflammation including infiltration of leucocytes.2 The inflammatory reaction in the liver has been clearly shown to be critical for the development and aggravation of steatosis as well as for induction and progression of organ fibrosis.1 3 4 On a cellular level, the activation of liver-resident macrophages, traditionally called ‘Kupffer cells’, and the vast infiltration of monocytes into the injured liver have been identified as major pathogenic factors. Since this pool of hepatic macrophages releases essential proinflammatory cytokines (eg, tumour necrosis factor α (TNFα)) it thereby promotes hepatocellular stress responses and lipid accumulation. Moreover, hepatic macrophages produce profibrogenic mediators (eg, transforming growth factor β (TGFβ)) and can directly activate collagen-producing hepatic stellate cells, thus linking inflammation to aberrant wound healing in the liver.5–7
On a molecular level, compelling evidence exists that macrophage infiltration into the liver is primarily controlled by C–C chemokine receptor 2 (CCR2) and its main ligand CCL2, monocyte chemoattractant protein-1 (MCP-1), in mice and men.6–10 In the injured liver, MCP-1 is synthesised by activated stellate cells, hepatocytes, macrophages and endothelial cells,4 5 8 thereby representing a central and redundantly activated pathway for liver inflammation. Functionally, CCR2- or MCP-1-deficient mice were shown to be protected from liver fibrosis in experimental models.6 8 9 11 In patients with fibrosis and cirrhosis, increased hepatic MCP-1 expression levels as well as CCR2-dependent macrophage infiltration have been described.7 12 13 Furthermore, MCP-1 appears to be particularly upregulated in patients with NASH, both intrahepatically and systemically.14
We herein conducted experiments to test whether pharmacological inhibition of hepatic macrophage infiltration via blocking MCP-1 could ameliorate liver damage, steatosis and fibrosis in rodent liver injury models. We therefore used an MCP-1-inhibiting compound that is based on a so-called Spiegelmer. Spiegelmers are short, L-enantiomeric RNA molecules that can be developed to specifically bind and inhibit a target of interest conceptually similar to monoclonal antibodies. In contrast to aptamers, which are composed of natural D-nucleotides or their respective derivates, Spiegelmers are nuclease resistant and therefore biostable as well as immunologically inert through the use of non-natural L-nucleotides.15 By modifying Spiegelmers with large inert moieties such as polyethylene glycol (PEG), their half-life in the bloodstream can be increased to allow even long-term therapeutic approaches.16
The Spiegelmer employed in this study, mNOX-E36, binds with high affinity to murine MCP-1 and inhibits its biological effects at low nanomolar concentrations. As such, mNOX-E36 has been shown to be active as a therapeutic agent against macrophage-mediated diabetic nephrosclerosis in various rodent models.17 18 Its human equivalent, NOX-E36, is currently being tested in patients type II diabetes in a phase Ib clinic trial (http://www.ClinicalTrials.gov number NCT01085292). In order to assess the therapeutic potential of MCP-1 inhibition in chronic liver diseases, we investigated the effects of specific pharmacological MCP-1 inhibition with mNOX-E36 in experimental mouse models of toxic (carbon tetrachloride (CCl4)) and metabolic methionine–choline-deficient (MCD) diet liver injury, respectively.
Materials and methods
Spiegelmers
The MCP-1-inhibiting L-RNA Spiegelmer mNOX-E36 (40 nucleotide L-RNA oligonucleotide; 5′-GGCGACAUUGGUUGGGCAUGAGGCGAGGCCCUUUGAUGAAUCCGCGGCCA-3′) and the respective non-functional control molecule revmNOX-E36 (composed of the reverse nucleotide sequence, both conjugated at their 3′ ends with Y-shaped 40 kDa PEG) were synthesised at NOXXON Pharma AG (Berlin, Germany). mNOX-E36 binds specifically to murine MCP-1 (CCL2) and inhibits the biological effects of MCP-1 in vitro at low nanomolar concentrations.17 19 The molecules were administered subcutaneously three times per week at a dose of 20 mg/kg body weight.17 19 mNOX-E36 was either administered throughout the injury model or during the last 2 weeks in the MCD diet model, as indicated.
Migration assay
Total bone marrow was isolated from untreated mice and subjected to red cell lysis using Pharmlyse (BD, Franklin Lakes, NJ USA). CCL2-dependent migration assays (eBioscience, 100 ng/ml) were performed in the presence or absence of mNOX-E36 through a semipermeable migration membrane (pore size 5 μm, Millipore, Billerica, MA USA) under sterile cell culture conditions in Iscove's modified Dulbecco's medium. The migrated population was analysed by fluorescence-activated cell sorting (FACS).
Mice
C57BL/6 wild-type (WT) and CCR2−/− mice (backcrossed to the C57BL/6 background for more than eight generations) were maintained in our colony.6 All mice were housed in a pathogen-free environment. All experiments were performed with male animals at 6–8 weeks of age under ethical conditions approved by the appropriate authorities according to German legal requirements.
Induction of acute or chronic liver injury
Mice received 0.6 ml/kg body weight CCl4 (Merck, Darmstadt, Germany) mixed with corn oil intraperitoneally and were killed at the indicated time points. For induction of liver fibrosis, CCl4 was injected twice weekly for 6 weeks. Mice were killed 48 h after the last injection. As controls, animals received the same volume of tracer (corn oil) intraperitoneally. For induction of steatohepatitis and fibrosis, mice were fed an MCD diet for 8 weeks (MP Biomedicals, Cat. no. 390439, Solon, OH USA).
Analysis of blood, bone marrow and intrahepatic leucocytes
Whole blood was drawn from heart puncture, and bone marrow was isolated from excised femurs. Cells were subjected to red cell lysis using Pharmlyse (BD), washed twice with Dulbecco's modified Eagle's medium (DMEM) containing 5 mM EDTA and 0.5% bovine serum albumin (BSA), and then stained with antibodies. For flow-cytometric analysis of intrahepatic leucocytes, livers were perfused with 20–40 ml of phosphate-buffered saline (PBS), minced with scissors and subsequently digested for 30 min with collagenase type IV (Worthington, Lakewood, NJ USA) at 37°C. Digested extracts were pressed through 70 μm cell strainers to achieve single cell suspensions. A small aliquot was stained with CD45 to assess the relative amount of intrahepatic leucocytes (CD45+) among all liver cells. The remaining liver single cell suspension was subjected to density gradient centrifugation (LSM-1077, PAA Laboratories, Pasching, Austria) at 2000 rpm for 20 min at 25°C. Leucocytes were collected from the interface after centrifugation, washed twice with Hank's balanced salt solution containing 2% BSA and 0.1 mM EDTA, and subjected to FACS.6 20
Flow cytometry
Six-colour staining was conducted using combinations of the following monoclonal antibodies: F4/80 (Serotec, Raleigh, NC USA), CD115, CD4, CD11c, CD11b (all eBioscience, San Diego, CA USA), CD45, Gr1/Ly6C, Ly6G, CD19, CD25, NK1.1, CD8 and CD3 (all BD). Dead cells were excluded by Hoechst 33 258 dye (Sigma-Aldrich, St Louis, USA). Flow-cytometric analysis was performed on a FACS-Canto (BD) and analysed with FlowJo (Tree Star, Ashland, OR USA).
Liver enzymes, histology and immunohistochemistry
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity (UV test at 37°C) were measured in serum (Roche Modular preanalytics system, Rotkreuz, Switzerland). Conventional H&E, Sirius red and Oil red O stainings were performed according to standard protocols.21 Sirius red-stained pictures were analysed by the area fraction quantification program in a blinded fashion (ImageJ). Liver sections from fixed paraffin blocks were immunohistochemically stained according to standard procedures using the following antibodies: anti-mouse F4/80 (Serotec) and CD45 (BD). Absolute counts of CD45+ cells (leucocytes) per high-power field of stained liver sections were manually assessed by an experienced pathologist in a blinded fashion.
Real-time gene expression analysis
Livers were harvested and snap-frozen in liquid nitrogen. RNA was purified from frozen liver samples by pegGOLD (peqLab, Erlangen, Germany), and cDNA was generated from 1 μg of RNA using a cDNA synthesis kit (Roche). Quantitative real-time PCR (qPCR) was performed using SYBR Green Reagent (Invitrogen, Carlsbad, CA USA).22 Reactions were done twice in triplicate, and β-actin values were used to normalise gene expression.23 Primer sequences are available upon request.
Hydroxyproline, cytokine and triglyceride measurements
The hepatic hydroxyproline content (reflecting total collagen) was measured as described.6 MCP-1, interleukin 6 (IL-6), interferon γ (IFNγ) and TNFα concentrations were measured from protein extracts of liver by ELISA (eBiosciences). Protein content was quantified by a photometric assay (Biorad, Hercules, CA USA). The intrahepatic triglyceride content was measured by TG liquicolor mono (Human Diagnostics, Wiesbaden, Germany) according to the manufacturer's instructions from homogenised snap-frozen liver samples.
Statistical analysis
All data are presented as the mean±SD. Differences between groups were assessed by two-tailed unpaired Student t test (GraphPad prism, La Jolla, CA USA).
Results
mNOX-E36 efficiently inhibits mouse monocyte migration in vitro
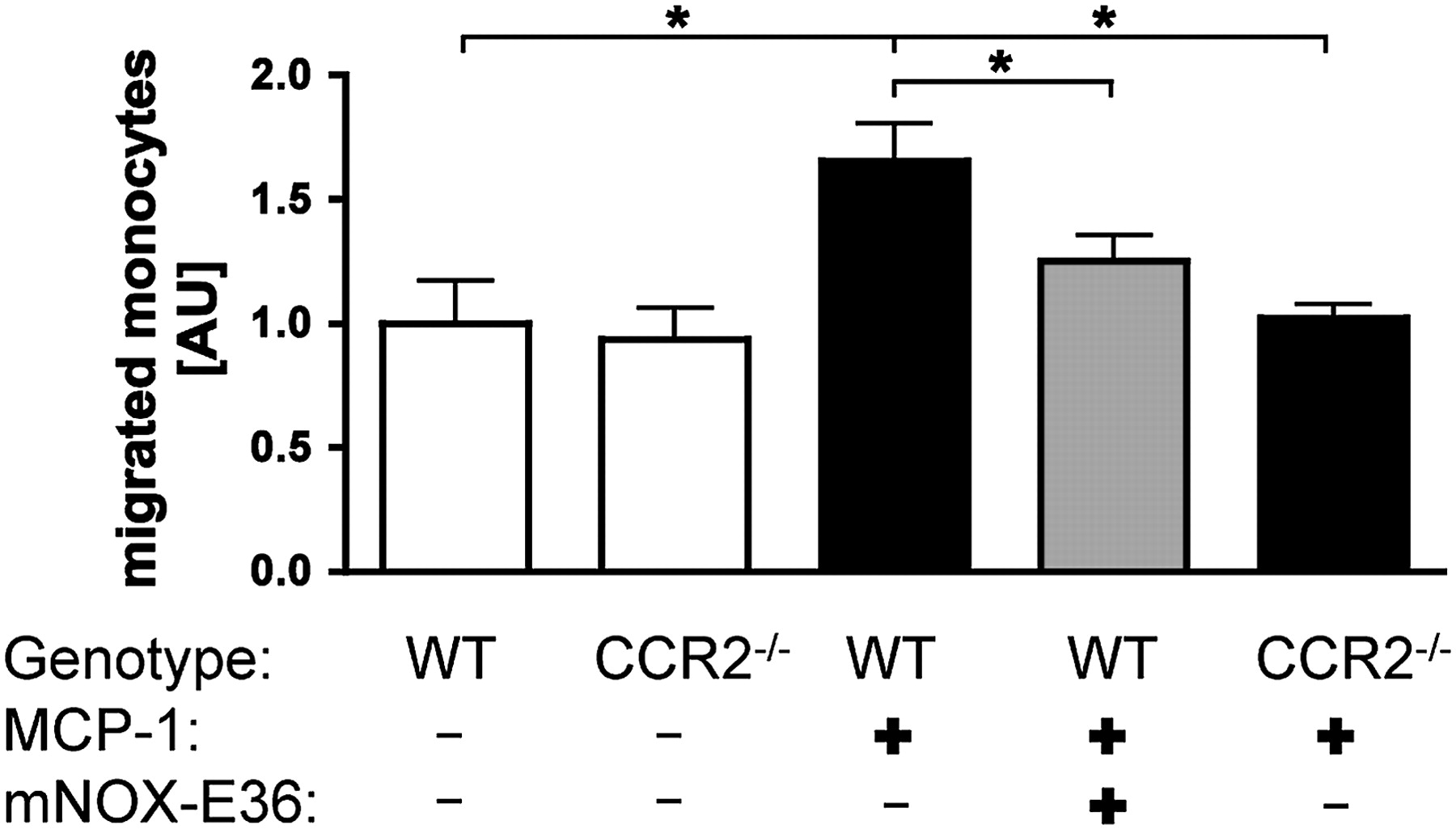
Recent studies using CCR2−/− mice demonstrated that chemokine receptor CCR2 and its ligand MCP-1/CCL2 promote the infiltration of inflammatory Gr1+ (Ly6C+) monocytes into the liver.6 8 9 Subsequently, these monocytes were found to exert proinflammatory along with profibrogenic actions by the release of distinct cytokines and direct interactions with hepatic stellate cells. Thus, inhibition of hepatic monocyte infiltration via blocking MCP-1 could represent a promising therapeutic approach during fibrogenesis. The main mechanism of MCP-1 action is likely to promote exit of Gr1+ monocytes from the bone marrow into the blood.6 24 25 In order to elucidate the possibility of pharmacological inhibition of monocyte migration by the MCP-1 antagonist mNOX-E36, bone marrow cells from C57BL/6 WT mice were first investigated in a chemotaxis experiment. Simultaneous incubation of cells with mNOX-E36 efficiently and specifically inhibited the MCP-1-dependent migration of bone marrow monocytes in a transwell assay (figure 1). The inhibition of MCP-1 via mNOX-E36 restrained monocyte migration to a similar extent as in CCR2−/− cells. Other leucocyte populations were not affected by mNOX-E36 (data not shown). This in vitro efficacy prompted us to assess whether mNOX-E36 is able to inhibit hepatic monocyte accumulation in liver injury models in vivo.

mNOX-E36 inhibits monocyte chemoattractant protein-1 (MCP-1, CCL2)-dependent migration of monocytes in vitro. Monocytes were isolated from bone marrow of either wild-type (WT) or chemokine receptor C–C chemokine receptor 2-deficient (CCR2−/−) mice. Migration of monocytes (defined as live CD11b+Ly6G−CD45+ cells) towards a gradient of 100 ng/ml recombinant murine CCL2 (MCP-1) was quantified after 4 h. Migration of unstimulated WT monocytes was set to 1. Data are expressed as mean±SD from three independent experiments. *p<0.05 (unpaired Student t test).
mNOX-E36 specifically inhibits hepatic macrophage infiltration in vivo upon acute liver injury
WT mice were challenged with a single dose of CCl4 intraperitoneally to induce acute toxic hepatic damage. CCl4 resulted in periportal necroses with maximal damage after 48 h (figure 2A). Correspondingly, highly elevated serum ALT activities (figure 2B) were measured. As assessed by FACS analysis (figure 2C) and immunohistochemistry (not shown) for the pan-leucocyte marker CD45 a severe accumulation of leucocytes in injured livers was also observed (figure 2C). Administration of mNOX-E36, however, efficiently reduced intrahepatic leucocyte infiltration, to a similar level to that seen in CCR2−/− mice (figure 2C).

mNOX-E36 specifically inhibits hepatic macrophage infiltration upon acute liver injury in vivo. Acute toxic liver injury was induced by 0.6 ml/kg carbon tetrachloride (CCl4) intraperitoneally in C57bl/6 wild-type (WT) mice. mNOX-E36 at 20 mg/kg was administered subcutaneoulsy simultaneously. Mice were analysed 48 h thereafter. (A) Conventional H&E staining of liver paraffin sections showed necrotic patches and progressive mononuclear infiltrates in the periportal regions after acute liver injury. (B) Hepatic injury was also assessed by serum alanine aminotransferase (ALT) levels. (C) Infiltration of leucocytes into the liver was quantified by fluorescence-activated cell sorting (FACS; percentage of CD45+ cells from total liver cells), revealing decreased counts in chemokine receptor C–C chemokine receptor 2-deficient (CCR2−/−) and mNOX-E36 treated mice. (D) FACS analysis of intrahepatic leucocytes, pregated on live (Hoechst dye−) CD45+Ly6G− cells, showing reduced numbers of CD11b+F4/80+ intrahepatic macrophages after mNOX-E36 injection and in CCR2−/− mice. Left, representative FACS plot; right, quantitative analysis. (E) FACS analysis of leucocytes from peripheral blood analysing circulating monocytes (defined as CD11b+Ly6G− cells). (F) FACS analysis of leucocytes from bone marrow analysing monocytes (defined as CD11b+Ly6G− cells). All data are expressed as the mean±SD from three independent experiments, comprising n=8–10 for animals in the treatment and n=5 animals in the control groups. *p<0.05, **p<0.005, ***p<0.001 (unpaired Student t test).
Previous studies revealed that in the early phase of liver injury macrophages are the predominantly infiltrating immune cells.6 20 Therefore, we purified leucocytes from liver tissues of WT animals and characterised the distinct cell types by FACS analysis. Intrahepatic macrophages were defined as living (Hoechst dye-negative) CD45+ cells that stain positive for the myeloid marker CD11b, positive for the macrophage marker F4/80 and negative for the neutrophil marker Ly6G.6 While WT mice showed massively increased hepatic macrophages after CCl4 injection, these cells only mildly increased in livers of mNOX-E36-treated WT animals. Like CCR2−/− mice, mNOX-E36-treated WT mice displayed significantly reduced macrophage infiltration following acute liver injury (figure 2D). Importantly, other leucocyte subpopulations such as T cells (CD8+, CD4+) or B cells (CD19+) were not affected by mNOX-E36 treatment (see supplementary figure 1 online).
Besides characterising the subsets of leucocytes in the liver, we also analysed corresponding monocyte populations in blood and bone marrow compartments. Of note, the hepatic CD11b+F4/80+ macrophages also stained positive for Gr1 (Ly6C), thus representing a vast majority of infiltrating monocytes from the circulation.6 Not only accumulation of these monocytes in injured liver, but also monocyte emigration out of the bone marrow was found to be inhibited by mNOX-E36. This effect was shown by an increased pooling of CD11b+Ly6G– monocytes in blood and bone marrow compared with untreated mice (figure 2E,F). As described in previous studies,6 24 25 CCR2−/− mice also displayed retention of monocytes in the bone marrow. The pharmacological effects on monocyte migration were specific for mNOX-E36 because the control Spiegelmer rev-mNOX-E36 did not show these effects on monocytes (data not shown). Furthermore, CCR2−/− mice were not affected by mNOX-E36 with respect to liver injury, circulating or intrahepatic macrophages (see supplmentary figure 2 online), demonstrating that the effects of mNOX-E36 are mediated by specifically inhibiting MCP-1/CCR2-dependent mechanisms. The clear inhibition of hepatic macrophage accumulation upon injury by mNOX-E36 in an acute in vivo model built the basis to examine its application in a chronic setting.
mNOX-E36 specifically inhibits hepatic macrophage infiltration in vivo in two models of chronic liver injury
The infiltration of macrophages into the liver upon chronic injury has been convincingly linked to the progression of liver inflammation and fibrosis in mice and men.6 7 20 26 We therefore also investigated the therapeutic effects of mNOX-E36 in models of chronic liver damage. WT mice were challenged with CCl4 intraperitoneally twice a week for 6 weeks and simultaneously received mNOX-E36 (20 mg/kg) subcutaneously three times a week. CCl4-treated animals displayed severe toxic liver damage with increased serum ALT (figure 3A) and AST (data not shown), necrotic patches in histology and mononuclear infiltrates (figure 3B,C). The massive influx of leucocytes, especially macrophages, was visualised by immunohistological staining for CD45 and F4/80 (figure 3C). In line with the acute effects after single administration of CCl4 and mNOX-E36, long-term treatment of mice with mNOX-E36 attenuated leucocyte influx in chronically damaged liver comparable with CCR2−/− mice (figure 3B,C). Again, macrophages were the predominant leucocyte population in chronic injury, and (Ly6C+) macrophage accumulation was significantly reduced in mNOX-E36-treated and in CCR2−/− mice, as demonstrated by immunohistochemistry (figure 3C) and FACS analyses (figure 3D). As expected and in line with the data of the acute model, the inactive Spiegelmer control had no effect on the amount of intrahepatic macrophages in this chronic setting (data not shown). Parallel to the intrahepatic macrophages we investigated other immune cells such as T cells (CD8+, CD4+) or natural killer (NK) cells (NK1.1+), which were affected neither in the CCR2-knockout nor by the mNOX-E36 treatment (see supplmentary figure 3 online). Besides liver tissue we again analysed blood and bone marrow by FACS. In blood we detected, concordant with the CCR2−/− mice, lower levels of monocytes in the mNOX-E36-treated mice (figure 3E). CCR2−/− mice, but not mNOX-E36-treated WT animals. retained monocytes in the bone marrow (figure 3F).

mNOX-E36 specifically inhibits hepatic macrophage infiltration upon chronic carbon tetrachloride (CCl4)-induced toxic injury in vivo. Chronic toxic liver injury was induced by 0.6 ml/kg CCl4 intraperitoneally twice weekly in C57bl/6 wild-type (WT) mice. mNOX-E36 at 20 mg/kg was administered subcutaneously three times per week. Mice were analysed after 6 weeks of repetitive CCl4 treatment, 48 h after the last injection. (A) Hepatic injury was assessed by serum alanine aminotransferase (ALT) levels. (B) Infiltration of leucocytes into the liver was quantified by fluorescence-activated cell sorting (FACS; percentage of CD45+ cells from total liver cells), revealing decreased intrahepatic leucocytes in chemokine receptor C–C chemokine receptor 2-deficient (CCR2−/−) and mNOX-E36-treated mice. (C) Representative immunohistochemical stainings for total leucocytes (CD45) and macrophages (F4/80) on liver paraffin sections from different experimental conditions. (D) FACS analysis of intrahepatic leucocytes, pregated on live (Hoechst dye−) CD45+Ly6G− cells, showing reduced numbers of CD11b+F4/80+ intrahepatic macrophages after mNOX-E36 injection and in CCR2−/− mice. Left, representative FACS plot, right, quantitative analysis. (E) FACS analysis from peripheral blood analysing circulating monocytes (defined as CD11b+Ly6G− cells). (F) FACS analysis of monocytes (defined as CD11b+Ly6G− cells) from bone marrow. All data are expressed as the mean±SD from three independent experiments, comprising n=10–13 for animals in the treatment and n=5–7 animals in the control groups. *p<0.05, **p<0.005, ***p<0.001 (unpaired Student t test).
In order to exclude that these results had been confounded by model-specific effects, we employed a second model of liver injury. Mice were fed an MCD diet for 8 weeks, resulting in severe steatohepatitis at a macroscopic and microscopic level (figure 4A). Mice were severely ill with substantial weight loss (figure 4B) and elevated ALT (figure 4C) and AST (data not shown) levels in comparison with mice fed a control diet. Liver histology showed a diffuse infiltration with leucocytes, apoptotic areas and a massive fatty degeneration (figure 4A). By immunohistochemistry and FACS analysis, we again found decreased numbers of intrahepatic leucocytes as well as macrophages in CCR2−/− and mNOX-E36-treated WT animals upon chronic metabolic injury (figure 4D). Other intrahepatic leucocyte populations were not affected in the MCD diet model either (see supplementary figure 4 online). Comparable with the toxic injury model, the total numbers of blood monocytes were decreased for CCR2−/− and mNOX-E36-treated mice (figure 4E), whereas only in CCR2−/− mice was the monocyte population retained in the bone marrow (figure 4F). Of note, lack of CCR2 or MCP-1 inhibition by mNOX-E36 also affected subpopulations of circulating blood monocytes, because Gr1hi monocytes were reduced in comparison with their Gr1low-expressing counterparts (see supplementary figure 4F online). Collectively, these data demonstrated that hepatic accumulation of macrophages upon chronic liver injury can be effectively attenuated by mNOX-E36 in vivo.

mNOX-E36 specifically inhibits hepatic macrophage infiltration upon chronic methionine–choline-deficient (MCD) diet-induced metabolic injury in vivo. C57bl/6 wild-type (WT) mice were fed an MCD diet for 8 weeks. mNOX-E36 at 20 mg/kg was administered subcutaneously three times per week. (A) Representative macroscopic pictures of the livers (scale marks indicate 1 cm) and corresponding H&E staining of liver paraffin sections. (B) Change of body weight from baseline after 8 weeks of MCD diet in the different treatment groups. (C) Hepatic injury was assessed by serum alanine aminotransferase (ALT) levels. (D) Fluorescence-activated cell sorting (FACS) analysis of intrahepatic leucocytes, presented as either total leucocytes (defined as live CD45+ cells, left) or macrophages (defined as live CD45+CD11b+F4/80+Ly6G− cells, right). (E) FACS analysis of leucocytes from peripheral blood analysing circulating monocytes (defined as CD11b+Ly6G− cells). (F) FACS analysis of monocytes (defined as CD11b+Ly6G− cells) from bone marrow. All data are expressed as the mean±SD from three independent experiments, comprising n=10–12 for animals in the treatment and n=5–7 animals in the control groups. *p<0.05, **p<0.005, ***p<0.001 (unpaired Student t test).
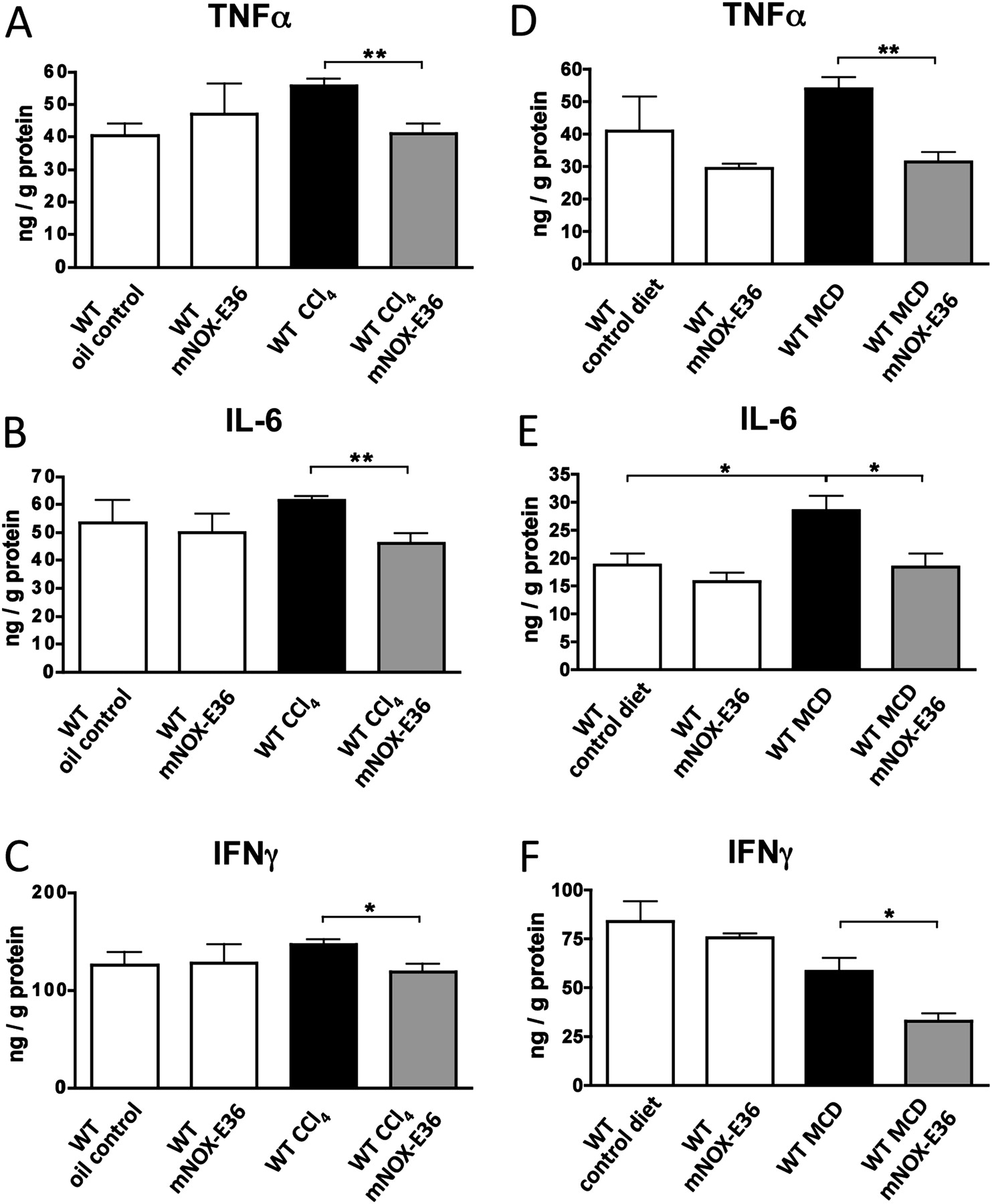
Reduction of liver-infiltrating macrophages via mNOX-E36 affects intrahepatic proinflammatory cytokines upon injury
We next investigated the consequences of blocking hepatic macrophage accumulation via mNOX-E36 in the liver. An important function of hepatic macrophages during disease progression is the secretion of proinflammatory cytokines.7 20 27 Therefore, cytokines were measured from total liver protein lysates by ELISA in both injury models. Animals that had received long-term treatment with mNOX-E36 had lower intrahepatic concentrations of TNFα, IL-6 and IFNγ upon CCl4- (figure 5A–C) or MCD diet-induced chronic liver injury (figure 5D–F), which is in line with the reduced numbers of intrahepatic macrophages. Gene expression analysis by qPCR confirmed these observations (data not shown). As expected, mNOX-E36-treated animals showed much higher levels of MCP-1 in the liver by ELISA (data not shown), because MCP-1 that is functionally antagonised by mNOX-E36 is still detected by the ELISA.17 Altogether, the clear reduction of proinflammatory cytokines in livers of mNOX-E36-treated animals indicated that pharmacological inhibition of MCP-1 might be capable of limiting chronic liver injury in vivo.
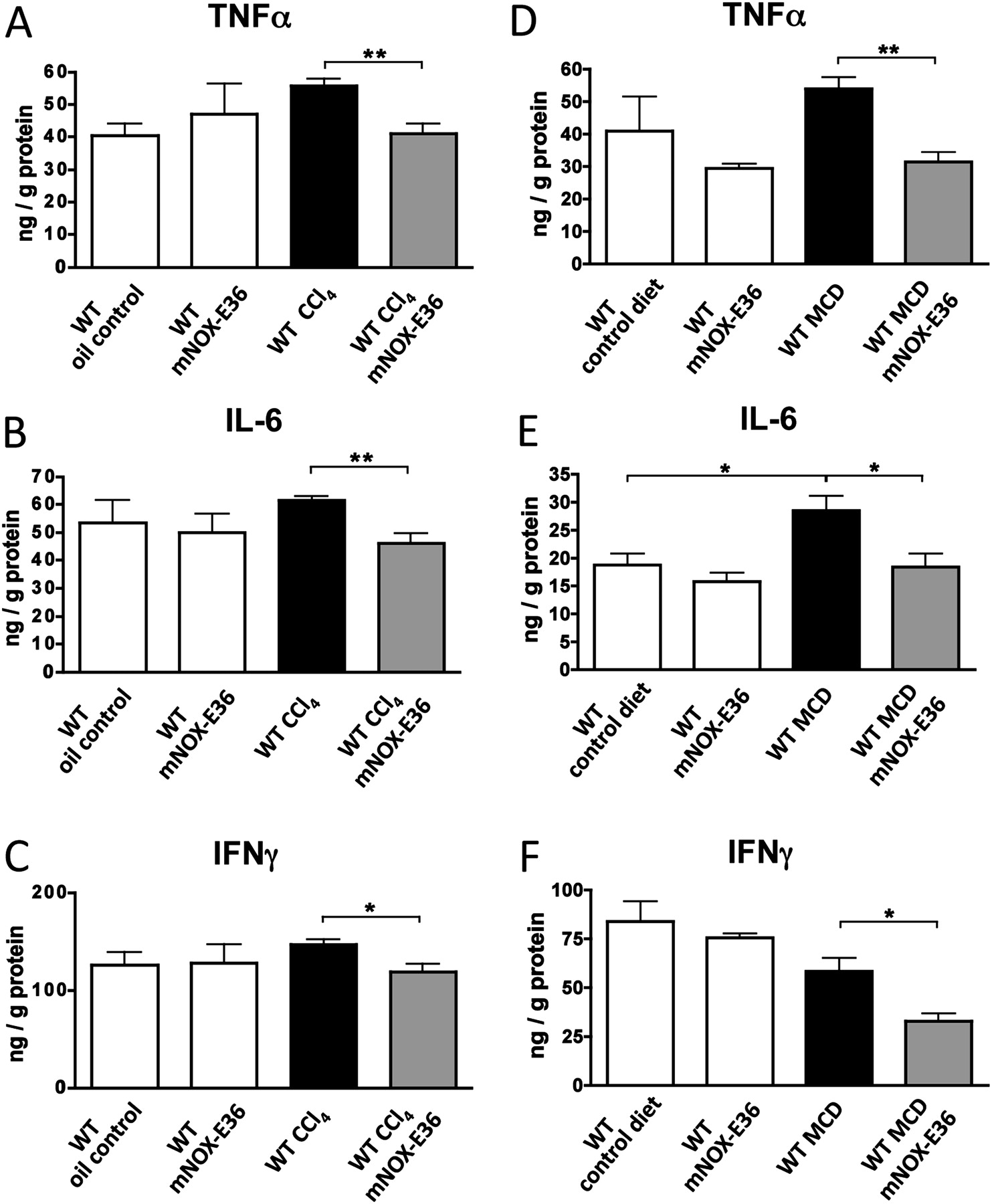
mNOX-E36 administration affects intrahepatic proinflammatory cytokines upon injury. C57bl/6 wild-type (WT) mice were subjected to 6 weeks of carbon tetrachloride (CCl4) injury (A–C) or to 8 weeks of a methionine–choline-deficient (MCD) diet (D–F) and treated long term with mNOX-E36 (3× per week). Cytokines were measured from total liver protein extracts by ELISA. All data are expressed as the mean±SD from three independent experiments, comprising n=10–12 for animals in the treatment and n=5–7 animals in the control groups. *p<0.05, **p<0.005 (unpaired Student t test). IFNγ, interferon γ; IL-6, interleukin 6; TNFα, tumour necrosis factor α.
Steatosis development, but not fibrosis progression, is significantly attenuated in mNOX-E36-treated mice
We next analysed whether pharmacological inhibition of MCP-1 might represent a successful therapeutic approach to limit disease progression in chronic liver injury models. We thus assessed liver fibrosis progression in both models by quantifying collagen deposition in Sirius red staining as well as measuring the hepatic hydroxyproline content, an amino acid that is abundantly found in collagen fibres (figure 6). While CCR2−/− mice showed an attenuated development of liver fibrosis in the chronic CCl4 model (figure 6A–C), as anticipated from prior studies,6 8 9 and also after an MCD diet (figure 6D–F), treatment with mNOX-E36 did not significantly affect hepatic fibrogenesis in either experimental model.

mNOX-E36 administration does not significantly alter hepatic fibrosis progression in two independent experimental models. C57bl/6 wild-type (WT) mice were subjected to 6 weeks of carbon tetrachloride (CCl4) injury (A–C) or to 8 weeks of methionine–choline-deficient (MCD) diet (D–F) and treated long-term with mNOX-E36 (3× per week). Collagen deposition in the liver was visualised by Sirius red staining (A, D), showing bridging fibrosis after CCl4 administration (A) and scattered collagen in steatotic livers after an MCD diet (D). Histological collagen deposition was quantified in a blinded, computer-based fashion (B, E). Hydroxyproline content of the livers was quantified as an additional measure of total hepatic collagen (C, F). Collectively, the data demonstrated attenuated fibrosis development in chemokine receptor C–C chemokine receptor 2-deficient (CCR2−/−) mice, but no significant effect of mNOX-E36. All data are expressed as the mean±SD from three independent experiments, comprising n=10–13 for animals in the treatment and n=5–7 animals in the control groups. *p<0.05, **p<0.005, ***p<0.001 (unpaired Student t test).
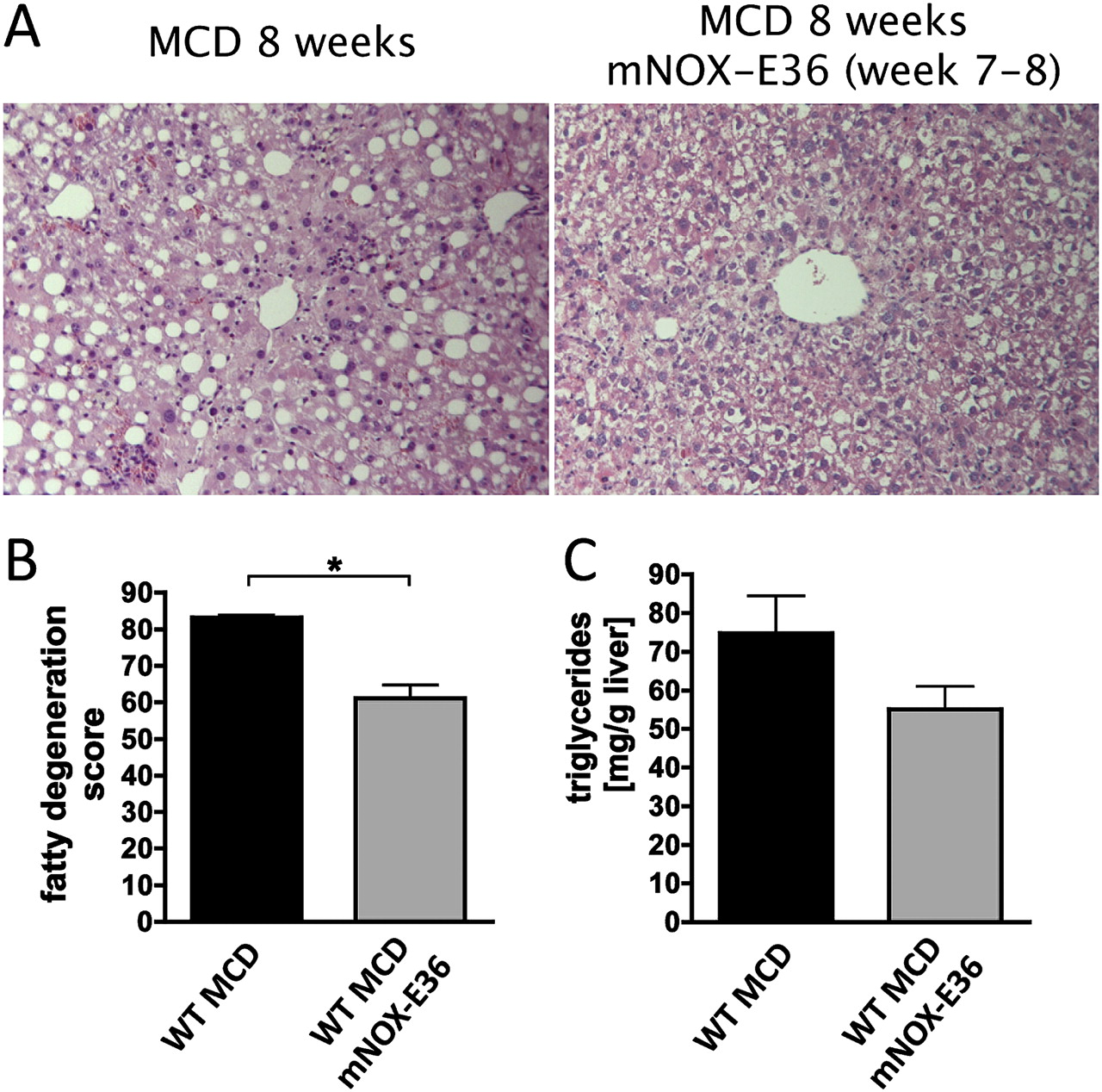
Several recent observations have linked the progression of steatohepatitis to the stimulation of TNF-dependent signalling pathways in hepatocytes upon injury.28 29 Thus, we hypothesised that the reduction of intrahepatic macrophages and threir associated proinflammatory cytokines might reduce the development of steatosis in vivo. Indeed, fatty degeneration of the liver was attenuated in mice fed an MCD diet during mNOX-E36 treatment as demonstrated by reduced steatosis by Oil red O staining (figure 7A) as well as histopathological criteria scored by a blinded and experienced pathologist (figure 7B). Subsequently, hepatic triglyceride content was significantly decreased in mNOX-E36-treated animals in the MCD diet model (figure 7C). Similar findings were obtained in CCl4-treated mice as well, although steatosis was much less prominent in this toxic model compared with the model with the MCD diet (data not shown). It has been reported previously that inflammatory cytokines from macrophages may induce the expression of important lipogenesis genes in injured liver.30 In line with the attenuated steatosis development, expression of the protective transcription factor PPARγ (peroxisome proliferator-activated receptor γ) was induced, whereas PEPCK (phosphoenolpyruvate carboxykinase), an important driver of hepatic gluconeogenesis, was reduced in mNOX-E36-treated animals in the MCD diet model (figure 7D,E). Thus, our results demonstrated that steatosis progression is closely associated with MCP-1-dependent macrophage recruitment and subsequent inflammation.

mNOX-E36 protects from steatosis development in experimental liver injury. C57bl/6 wild-type (WT) mice were fed a methionine–choline-deficient (MCD) diet for 8 weeks. mNOX-E36 at 20 mg/kg was administered subcutaneously three times per week. (A) Oil red O staining of liver cryosections showed decreased levels of positive stained intracellular fat vacuoles in mNOX-E36-treated mice. (B) Pathological scoring of liver histology by a blinded pathologist showed attenuated fatty degeneration in mNOX-E36-injected mice. (C) Quantification of triglycerides from total liver lysates revealed reduced triglyceride content in murine liver after mNOX-E36 treatment. (D, E) Gene expression analysis from whole liver by quantitative real-time PCR showed induced hepatic peroxisome proliferator-activated receptor γ (PPARγ) and reduced phosphoenolpyruvate carboxykinase (PEPCK) mRNA levels in mNOX-E36-injected mice. All data are expressed as the mean±SD from three independent experiments, comprising n=10–12 for animals in the treatment and n=5–7 animals in the control groups. *p<0.05, **p<0.005, ***p<0.001 (unpaired Student t test).
mNOX-E36 also ameliorates steatosis development upon therapeutic application
In order to test the therapeutic potential of mNOX-E36 to treat ongoing fatty liver degeneration, mice were kept on an MCD diet for 6 weeks without treatment, which induced severe steatohepatitis (not shown). While the MCD diet was continued, mNOX-E36 was administered during the last 2 weeks of injury. Clearly, mNOX-E36 treatment in this therapeutic setting reduced hepatic steatosis, as intrahepatic lipid vacuoles (figure 8A) and the histological fatty degeneration score (figure 8B) improved upon MCP-1 inhibition. Moreover, the hepatic triglyceride content showed a trend towards lower levels in the mNOX-36-treated animals (figure 8C). These data indicated that inhibition of MCP-1 might be a promising therapeutic strategy even in ongoing or established steatohepatitis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
mNOX-E36 ameliorates hepatic steatosis in a therapeutic setting. C57bl/6 wild-type (WT) mice were fed a methionine–choline-deficient (MCD) diet for 8 weeks. mNOX-E36 at 20 mg/kg was administered subcutaneously three times per week only during the last 2 weeks of the diet. (A) Conventional H&E staining of liver paraffin sections showed fatty degeneration after feeding the MCD diet. Treatment with mNOX-E36 resulted in lower amounts of cells with large fat vacuoles. (B) Pathological scoring of liver histology by a blinded pathologist showed attenuated fatty degeneration in mNOX-E36-injected mice. (C) Quantification of triglycerides from total liver lysates revealed a trend of reduced triglyceride content in murine liver after mNOX-E36 treatment. All data are expressed as the mean±SD with n=4 animals per group. *p<0.05 (unpaired Student t test).
Discussion
Chronic liver injury is accompanied by a prominent inflammatory response, which subsequently triggers hepatocytic stress responses (eg, apoptosis or steatosis) as well as wound healing reactions by non-parenchymal cells (ie, fibrosis). Hepatic inflammation is a highly complex process in which the increased expression of distinct CC and CXC chemokines and their respective receptors have been identified as major regulators of immune cell recruitment into the injured liver.4 31 Among these mediators, compelling evidence exists that the CCR2/CCL2 axis executes a predominant role in liver inflammation. Several independent studies highlighted the importance of chemokine receptor CCR2 and its main ligand MCP-1/CCL2 for monocyte/macrophage recruitment during experimental hepatic fibrosis.13 These data suggest that inhibition of either CCR2 or MCP-1 might have therapeutic potential in chronic liver diseases. In our study, we demonstrate that the specific MCP-1 inhibitor mNOX-E36 effectively reduces intrahepatic macrophage accumulation and ameliorates steatosis development upon toxic and metabolic liver damage in mice.
In general, the CCR2–CCL2 interaction is responsible for the directed migration of monocytes/macrophages to areas of injury and inflammation in many diseases.32 CCR2/CCL2-mediated actions are important for migration of monocytes from the bone marrow into the blood,6 23–25 but also from the blood to inflamed tissues.23 33 Thus, blocking CCL2/MCP-1, the main ligand for CCR2, appears to be an attractive (targeted) therapeutic approach in many inflammatory conditions. Pharmacological intervention with this axis in experimental models of obesity has already been demonstrated to reduce macrophage infiltration into adipose tissue and to improve insulin sensitivity.34–36
The MCP-1-specific Spiegelmer mNOX-E36 is an interesting pharmacological modality because it is highly effective at nanomolar concentrations, has favourable pharmacokinetic characteristics (suggesting efficient plasma levels at a once- or twice-weekly dosing schedule) and no significant immunogenicity in vivo.16–19 Our experiments demonstrated that the Spiegelmer mNOX-E36 is not only able to inhibit monocyte migration in vitro, but also effectively and specifically reduces the accumulation of intrahepatic macrophages upon acute and chronic liver injury in vivo. These effects were independent of the underlying cause of liver damage, as similar rates of hepatic macrophage reduction were seen in chronic toxic or metabolic injury models.
Macrophages in the liver were shown to exert functionally different actions, for instance perpetuation or resolution of hepatic inflammation, dependent on their activation, differentiation and likely influences from the local hepatic microenvironment.13 20 26 In this study, we focused on progressing chronic liver disease models. During disease progression, mNOX-E36 administration significantly reduced the protein levels of hepatic proinflammatory cytokines (TNFα, IL-6). These data suggested that mNOX-E36 primarily affected ‘classical’ M1-type differentiated macrophages.13 However, we cannot exclude that different macrophage subsets may be altered during regression or resolution of liver injury.9
Overall, treatment with mNOX-E36 ameliorated hepatic steatosis, but not fibrosis in two independent chronic injury models. Interestingly, similar data have been obtained with distinct knockout mice. While CCR2−/− mice were protected from fibrosis development in several independent studies.6 8 9 MCP-1-deficient animals also showed reduced fibrosis in a model of an MCD diet administered for 4 weeks.11 Given the clear link between macrophages and fibrosis progression,6 13 26 several factors may account for this observation. First, our murine models (especially in ‘slow-fibrosing’ C57bl/6 mice) investigated a rather early stage of fibrosis progression; possibly, long-term effects on fibrosis may have been overlooked in this experimental setting and may be apparent at more progressed disease. Secondly, the reduction of macrophages achieved by mNOX-E36 at this dose may not be suitable to suppress levels below a certain threshold, suggesting that higher doses should be evaluated in future studies or that mNOX-E36 should be combined with additional antagonists for alternative CCR2 ligands such as MCP-2 (CCL8), MCP-3 (CCL7) and MCP-5 (CCL12).24 Thirdly, blocking MCP-1 may sufficiently suppress infiltration of monocytes or macrophages into the liver, but not of other CCR2-expressing immune cells. Of note, a recent study identified haematopoietic and progenitor cells that traffic into injured liver via CCR2,33 which would be consistent with our finding that CCR2−/− mice differ substantially from the mNOX-E36-treated animals with respect to fibrosis development. Fourthly, mNOX-E36 primarily abolished typical M1-type macrophage functions such as TNF production; however, several lines of evidence indicate that alternatively activated macrophages (M2) may specifically exert profibrogenic actions, such as TGFβ synthesis or stellate cell activation.13
Importantly, treatment with mNOX-E36 significantly improved the development of hepatic steatosis upon chronic MCD diet-induced metabolic injury. This effect could also be reproduced when mNOX-E36 was administered therapeutically during ongoing liver injury. Our data emphasise that macrophages, in an MCP-1-dependent manner, interact with hepatocytes in the regulation of hepatic triglyceride storage. In fact, it has been previously reported that hepatic macrophages promote deposition of liver triglycerides by inflammatory cytokines such as TNF or IL-1β, which suppresses PPARα expression and activity.30 37 Of note, MCP-1 knockout mice also showed a trend towards lower hepatic steatosis and triglyceride deposition after 4 weeks of an MCD diet, but differences from WT mice did not reach significance in this short-term model.11 Our observation of reduced hepatic steatosis progression upon liver injury by mNOX-E36 administration is also in agreement with studies in which mice were fed a high fat diet; here, MCP-1 deficiency or CCR2 blocking also ameliorated hepatic steatosis and triglyceride content.35 36 In these studies, pro-inflammatory cytokines released by macrophages into the liver but also into other compartments such as adipose tissue were identified as crucial inducers of hepatic steatosis.35 36 Our data suggest that pharmacological inhibition of MCP-1 by mNOX-E36 should also be evaluated in high fat diet-induced steatosis models, as these models would in some aspects (obesity, slow progression, systemic inflammation and atherosclerosis) more closely resemble the human situation of patients with NASH.
The human equivalent of the MCP-1 inhibitor mNOX-E36 is currently being evaluated in clinical trials for the treatment of diabetic nephropathy. As patients with diabetes with metabolic syndrome almost always coincidentally display hepatic steatosis and have a highly increased risk of developing NASH,38 our study suggests that treatment with mNOX-E36 might have additional advantages for patients with diabetes by mediating antisteatotic and anti-inflammatory actions in the liver.
Acknowledgments
The authors thank Aline Müller for excellent technical assistance, Martin Humphrey for helpful discussions, and the NOXXON chemistry group for synthesising the Spiegelmer molecules.
References
Footnotes
Funding This work was supported by the German Research Foundation (DFG Ta434/2-1 to FT, SFB/TRR 57) and funding from NOXXON (Berlin, Germany). DE and SK are employees of NOXXON. The funders were not involved in data analysis or interpretation.
Competing interests DE and SK are employees of NOXXON Pharma AG who provided part of the funding for this study.
Provenance and peer review Not commissioned; externally peer reviewed.