Article Text

Abstract
Advances in sequencing technology and the development of metagenomic and bioinformatics methods have opened up new ways to investigate the 1014 microorganisms inhabiting the human gut. The gene composition of human gut microbiome in a large and deeply sequenced cohort highlighted an overall non-redundant genome size 150 times larger than the human genome. The in silico predictions based on metagenomic sequencing are now actively followed, compared and challenged using additional ‘omics’ technologies. Interactions between the microbiota and its host are of key interest in several pathologies and applying meta-omics to describe the human gut microbiome will give a better understanding of this crucial crosstalk at mucosal interfaces. Adding to the growing appreciation of the importance of the microbiome is the discovery that numerous phages, that is, viruses of prokaryotes infecting bacteria (bacteriophages) or archaea with a high host specificity, inhabit the human gut and impact microbial activity. In addition, gene exchanges within the gut microbiota have proved to be more frequent than anticipated. Taken together, these innovative exploratory technologies are expected to unravel new information networks critical for gut homeostasis and human health. Among the challenges faced, the in vivo validation of these networks, together with their integration into the prediction and prognosis of disease, may require further working hypothesis and collaborative efforts.
- Bacterial interactions
- genetics
- IBD
- mucus
- intestinal microbiology
- metagenomics
- carbohydrates
- anaerobic bacterial fermentation
- molecular biology
- mucins
- inflammatory diseases
- bacterial adherence
- short chain fatty acids
- IBD basic research
- intestinal microbiota
- Crohn's disease
- stool markers
- inflammatory bowel disease
- intestinal bacteria
- ribosomal RNA
- colonic microflora
- enteric bacterial microflora
- RNA expression
- gut inflammation
- intestinal bacteria
- immune response
- mucosal immunology
Statistics from Altmetric.com
- Bacterial interactions
- genetics
- IBD
- mucus
- intestinal microbiology
- metagenomics
- carbohydrates
- anaerobic bacterial fermentation
- molecular biology
- mucins
- inflammatory diseases
- bacterial adherence
- short chain fatty acids
- IBD basic research
- intestinal microbiota
- Crohn's disease
- stool markers
- inflammatory bowel disease
- intestinal bacteria
- ribosomal RNA
- colonic microflora
- enteric bacterial microflora
- RNA expression
- gut inflammation
- intestinal bacteria
- immune response
- mucosal immunology
Introduction
Shortly after the publication of the first human genome draft in 2001 it became obvious that a complete understanding of the human biology would only be fully assessed by combining the analysis of the host and its surrounding environment.1 The human gastrointestinal tract hosts more than 100 trillion bacteria and archaea which together make up the gut microbiota. Archaea are prokaryotes and represent a third domain between bacteria and eukaryotic organisms. First described from extreme environments, archaea exist in a broad range of habitats and may contribute up to 20% of the Earth's biomass. Most of the cultivable species are members of two phyla, the Euryarchaeota and Crenarchaeota. In gut ecosystems, they are methanogenic, from the Euryarchaota phylum. Even though the amount of bacteria in the human gut outnumber human cells by a factor of 10,2 ,3 some finely tuned mechanisms allow these microorganisms to colonise and survive within the host in a commensalism relationship.4 This tolerance phenomenon is facilitated through physical separation of bacteria and host cells via the mucus layer, but also through modifications of antigenic moieties of the microbiota components to reduce their immunogenic properties and by direct modulation of localised host immune responses.5–7
The human gut microbiota can be considered an organ within an organ8 that co-evolved with humans to achieve a symbiotic relationship leading to physiological homeostasis.9 The human host provides a nutrient-rich environment and the microbiota provides indispensable functions that humans cannot exert themselves, such as the production of some vitamins, digestion of complex polysaccharides10 and the shaping of the immunological environment.11 ,12 Commensal bacteria influence the normal development and function of the mucosal immune system, the induction of IgA and epithelial barrier tightening. Through the production of short-chain fatty acids, mainly acetate, propionate and butyrate, resident bacteria positively influence intestinal epithelial cell proliferation and differentiation, and also mediate various metabolic effects.13
Development of cultivation-independent methods based on 16S small subunit (SSU) rRNA gene sequence analysis rapidly expanded our knowledge about the diversity of the gastrointestinal tract microbiota. Only a decade after their first application to the human gut,14 the number of detected gastrointestinal tract phylotypes using molecular techniques has by far outnumbered the cultivated gut species. From more than 1200 microbes described, only 12% were recovered by application of both molecular and cultivation-based approaches, while the vast majority (∼75%) were detected solely through SSU rRNA sequencing.15
Chaotic in the early stages of human life,16 ,17 the assembly of the human gut microbiota remains globally stable over time in healthy conditions in the absence of perturbation.18 ,19 The average total number of bacterial species was estimated to be close to 1000 per individual whereas 10 000 to 40 000 are predicted for the whole microbiota population.20 ,21 The restricted number of phyla in comparison to other ecosystems (four out of 50 existing phyla) has suggested a tight co-evolutionary history between the host and its microbiota.22 ,23 This co-evolution is also observed at the species level and has led to the definition of a phylogenetic core composed of 66 highly prevalent and abundant species.21 Two per cent of one person's microbial species are shared by at least 50% of the population.
Remarkably, shifts in the bacterial makeup of the human gut microbiota have been associated with digestive tract dysfunctions such as inflammatory bowel disease (IBD), irritable bowel syndrome (IBS) and obesity.20 ,24–31 More than 10 years ago, the concept of dysbiosis or unbalanced composition of the intestinal microbiota, was introduced in the IBD research field.32 Although an impressive list of documented microbial alterations in patients with IBD has recently been reviewed,33 the original question of whether dysbiosis is just a secondary phenomenon in IBD or truly causal remains.34
Further insights into the human gut ecosystem are needed to comprehend the exact role of microbiota in health and disease. Because most of the bacteria inhabiting the gut are uncultivable, their functions cannot be inferred from composition data. Knowing which microbes are there is not sufficient. Essential questions are ‘What is the genetic potential of the non-cultured bacterial fraction of the gut microbiota?’ and ‘What are these microbes really doing?’. Meta-omics aims to answer these questions. Metagenomics, which is based on whole microbiome genomic content, has started to put forward the microbial functionalities embedded into the human gut microbiota. Among the key outputs, this review addresses the total number of encoding genes per person or as a whole, enterotypes definition and global functional properties.
Metagenomics: towards a better understanding of the human gut microbiome
Metagenomics was first described in 1998 by Handelsman and Rodon.35 ,36 It was defined as analysis of the collective genomes that are present in a defined environment or ecosystem, hence giving insight into functions of non-cultivated bacteria. It was first applied to the analysis of aquatic and soil ecosystems. The first golden standard for metagenomics was the use of metagenomic libraries. Large DNA insert metagenomic libraries were constructed using fosmids, cosmids or bacterial artificial chromosomes with single, low copy or copy control vectors. Total microbial DNA was extracted, separated in large DNA fragments using pulse-field gel electrophoresis before being cloned in host bacteria to generate a screening library.
The emergence of next generation sequencing technologies,37 such as pyrosequencing, SOLiD (‘Sequencing by oligonucleotide ligation and detection’) or Illumina led to the development of sequencing projects covering a broader fraction of the microbial diversity present in the original sample. It then became possible to sequence the inserts of clones of interest from metagenomic libraries rapidly and in a cost-effective manner but also to directly shotgun sequence the metagenomic DNA content of an ecosystem. The bioinformatics analysis of a sequence, so called annotation, from open reading frames finding to similarity searches in databases, enables researchers to identify major functions and genes that encode enzymes of clinical or industrial interest. The complete genome of particularly abundant microorganisms can also be reconstructed,38 ,39 providing a more comprehensive view of their biological potential (figure 1).

Main metagenomics applications, from the metagenomic libraries construction and screening, until next generation sequencing, gene count and genome reconstruction. Metagenomics libraries are screened for a set of bioactivities ranging from bacteria–food metabolic activities (such as glycoside hydrolases production or butyrate production) to bacteria–bacteria interactions (ie, quorum sensing or antimicrobials activities). These bioactivities are further described by inserts sequencing and annotation. However, metagenomic DNA can be directly sequenced. Following contigs assembly, analyses ranging from bacterial genome reconstruction to systems ecology can be applied. From sequences data, quantitative metagenomics can also be applied to unravel the bacterial genes within an ecosystem. BAC, bacterial artificial chromosome; COS, cosmid; FOS, fosmid.
Nonetheless, the scale of these analyses needs to be adequate to permit the mapping of the complete human microbial metagenome, given the incredible abundance and complexity of the microbial populations found in the digestive tract. An average of 4.5 gigabases (Gb; ranging between 2 and 7.3 Gb) of sequence were generated for each faecal sample of the 1st MetaHIT study.40 Considering an average size of bacterial chromosomes of 4.5 megabases (Mb; ranging from 0.6 Mb to over 10 Mb), the gene catalogue was therefore equivalent to that of some 1000 bacterial species, encoding about 3364 non-redundant genes for each person's microbiome, capturing most of the novelty.
The first sequence-based characterisation was obtained from samples of two American healthy volunteers41 and showed a significant enrichment of the gut microbiome in metabolic pathways related to metabolism of glycans, amino acids and xenobiotics; methanogenesis; and 2-methyl-D-erythritol 4-phosphate pathway-mediated biosynthesis of vitamins and isoprenoids. Shortly after, the metagenomic analysis of 13 healthy Japanese volunteers42 confirmed, at functional levels, the chaotic picture drawn from previous compositional studies of unweaned infants' gut microbiome.17 Nevertheless, a set of 136 gene categories was found to be prevalent in infant-type microbiome, clearly directed towards carbohydrate transport and metabolism facilitating nutrient uptake from milk. The adult-type microbiome was found to be commonly enriched with 237 gene families, suggesting host–microbiota co-evolution towards functionalities favouring energy harvest from diet (carbohydrate metabolism) and bacterial competition (antimicrobial peptide transporters). In addition, genes encoding the biosynthesis of flagella and related to chemotaxis were depleted, suggesting that commensal microbes lowered their immunogenic properties to persist into the gut.
Finally, among the first human gut metagenomic libraries were two large insert libraries generated from pools of faecal samples from six healthy people and six patients with Crohn's disease.29 ,43 Analysis of the corresponding gene content at the phylogenetic level indicated, as expected from previous works, broad species diversity in healthy people and, inversely in the Crohn's disease (CD) faecal library, a markedly reduced species diversity belonging to the Firmicutes, mostly from the Clostridium leptum group.
Functional metagenomics: discovering new functions and re-evaluating microbial pathways
A challenging approach for metagenomic analysis is to identify clones bearing bacterial genes that express a function. Because 86% of bacterial genomic DNA encodes for coding open reading frames,44 clones bearing insert fragments of more than 1 kilobase can be screened for genes and pathways, applying functional metagenomics to the cultured and mostly uncultured bacteria. Although Escherichia coli is the most used host for functional metagenomics, other species including Streptomyces, Bacillus subtilis or Lactococcus lactis can be chosen to facilitate the heterologous expression of Gram-positive bacterial DNA.45 However, the taxonomic diversity of bacteria which genes could be expressed in E coli has been reported unexpectedly high.46
The first screenings of gut metagenomic libraries were applied to the catabolism of dietary fibres, which is of particular interest in human nutrition and health. Dietary fibres have been identified as a strong positive dietary factor in the prevention of obesity, diabetes, colorectal cancer and cardiovascular diseases (World Health Organization, Joint WHO/FAO Expert Consultation “Diet, Nutrition and the Prevention of Chronic Diseases” 2003, WHO Technical Report Series 916). Human gut bacteria produce a vast panel of carbohydrate active enzymes (CAZymes) to degrade components of dietary fibre into metabolisable monosaccharides and disaccharides. CAZymes study was nevertheless restricted to cultivated bacterial species. CAZymes diversity has been described in three metagenomics studies focused on the gut microbiome,41 ,47 ,48 revealing the presence of at least 81 families of glycoside hydrolases, making the human gut metagenome one of the richest sources of CAZymes.49 Tasse et al applied high-throughput functional screenings on human gut derived metagenomics clones (156 000 clones representing 5.5 Gb of DNA) and identified new CAZymes.50
Similarly, Jones and colleagues screened about 90 000 fosmid clones from the human gut metagenome (representing about 3.6 Gb bacterial DNA) for bile salt hydrolase activity.51 These functions were found to be present and enriched in all major gut microbial divisions, including archaea. A different strategy allowed the expression of metagenomic β-glucuronidase activity in β-glucuronidase-postive E coli fosmid libraries from healthy people and patients with CD.52 The study unravelled a new subfamily of β-glucuronidase, dominant in healthy adults and children, which may have specifically evolved to settle in the human gut microenvironment.
Even though the microbiota contributes to the homeostasis of the intestinal mucosa and the maturation of the immune system, the underlying cellular and molecular mechanisms driving the co-evolution and interplay between microbiota and host remain poorly understood. Since the intestinal epithelial cells are the first line in contact with microbes and have a key role in immune regulation and inflammatory pathways, the functional metagenomic approach was also validated for the identification of bacterial genes influencing the proliferation of HT-29 (colorectal carcinoma cell line) human intestinal epithelial cells.53 Using high-throughput screening and reporter gene technology in human epithelial cells, Lakhdari et al identified metagenomic clones activating or inhibiting the nuclear factor κB (NF-κB) signalling pathway.54 Among the bacterial genes modulating the NF-κB pathway were a putative lipoprotein of yet unknown function and an efflux ABC-type transport system, related to the Lol-D family of lipoprotein transporter complex.
Another application is the screening of plasmid encoding elements. The mobile genetic elements associated with the human gut microbiota (the mobile metagenome) reflect the co-evolution of host and microbe in this community. Genes involved in survival and persistence in the gastrointestinal tract, as well as activities that impact on host–microbe or microbe–microbe interactions, are encoded by the gut mobile metagenome (‘mobilome’). Applying the culture independent Transposon-aided capture (TRACA) system, Jones and Marchesi isolated novel plasmids from the human gut microbiota and detected genes enriched in the human gut compared with other ecosystems.55 Among the most prevalent functions identified was a putative RelBE toxin–antitoxin addiction module. Moreover, recent evidence from metagenomics indicated that horizontal gene transfer could happen between phylogenetically distant bacterial groups.42 ,50 ,51 Very recently, Smillie and colleagues decrypted the forces driving these exchanges and demonstrated that ecology is the main driver of gene exchange. For instance, bacteria with the same oxygen tolerance, that is, inhabiting the same body site and sharing ecological features, are actively exchanging genes.56
Finally, functional genomics can also provide calibrated assays designed to estimate the functionalities of cultivable microorganisms found to be over-represented or under-represented in several clinical conditions, such as inflammation, or to compare expression of probiotic strains for key functions related to human health.
Our other genome: highlights of the European MetaHIT project
In October 2005, an international meeting gathered together a panel of experts focusing on the challenges and feasibility of an exhaustive analysis of the human metagenome. As an outcome, the International Human Microbiome Consortium (IHMC) was created, the goals of which are to work under a common set of principles and policies to study and understand the role of the human microbiome in the maintenance of health and causation of disease, and to use that knowledge to improve the ability to prevent and treat disease. The Consortium's efforts have focused on generating a shared comprehensive data resource that enables investigators to characterise the relationship between the composition of the human microbiome and human health and disease (http://www.human-microbiome.org/). The IHMC's main research programmes are summarised in table 1.
International Human Microbiome Consortium (IHMC) main research programmes
Evidence for co-evolution and the high level of complexity of humans and their microbiome led to the hypothesis of functional redundancy in the intestinal microbiome. Different bacterial species share functional traits and the large amount of intestinal bacteria guarantees the presence of all mandatory functions, providing robustness to this ecosystem. The first extensive catalogue of microbial genes from the human gut, published by Qin and colleagues,40 described the large variety of traits provided by the intestinal microbiota and an overall metagenome per individual outnumbering by a factor of 150 the size of the human genome. This catalogue highlighted functions that are important for bacterial survival in the human intestines and the existence of a functional core, conserved in each individual of the cohort (n=124), despite a high inter-individual specificity of the intestinal microbiota. This functional core or minimal human gut metagenome can be viewed as a set of non-redundant bacterial genes necessary for the normal functioning of the gut ecosystem and encoded across phylogenetic boundaries. In the studied population, 38% of one individual's bacterial genes were shared by at least 50% of the cohort, highlighting a high level of functional similarities between individuals. This percentage fell to 9% when looking at genes shared by at least 80% of the community.
The depth of coverage and wealth of information provided by metagenomic sequencing recently enabled the discovery of distinct ‘types’ of gut composition in the human population. Through comparison of the phylogenetic and functional composition of the gut microbiota in three different international cohorts of 33, 85 and 166 individuals, Arumugam, Raes and colleagues found that nationality and the presumed similarity in genetic, ethnic and nutritional background was not reflected in gut composition similarity. Instead, they found three distinct clusters of gut composition (‘enterotypes’), characterised by the dominant genera and their co-occurring phylogenetic groups that separated the population.57 Analysis of the metagenomic data, based solely on the pathways that could be assigned to distinct functions, suggested that enterotypes differ at least in their capacity of vitamin production and metabolic dependencies of the dominant organisms. A recent 16S-based study evaluated the effect of dietary habits of 98 US individuals on their enterotype and found a positive correlation (false discovery rate <25%) between long-term diet (reported habitual diet over 1 year) and enterotypes. Protein and animal fat uptake was linked to the Bacteroides-dominated enterotype while a carbohydrate-rich diet was associated with the Prevotella-dominated enterotype. Short-term diet change and 10 days' dietary intervention affected the species composition but not the enterotype distribution.58 All in all, these studies still open up a wide range of questions about enterotypes and especially their stability over time in humans as a function of nutrition or clinical status.
Functions and dysfunctions of the gut microbiome
The latter studies issued from the MetaHIT programme led to an important breakthrough about functional gut microbiome impairment in inflammation-related diseases, particularly in IBD and obesity. The first study compared 124 individual metagenomes (healthy, overweight and obese individuals, and patients with IBD) and showed that only 25% of one's individual genes are shared with patients with IBD compared with 38% in healthy individuals.40 Remarkably, the number of non-redundant bacterial genes was significantly lower in patients with IBD (425 397 + 126 685, SD; n=25) than in healthy individuals (564 070 + 121 962, SD; n=99; p<10−6, one-tailed Student t test). Interestingly, functional genes or modules, and also gene counts, strongly correlate with an individual's clinical status (figure 2) and might represent diagnostic tools for numerous human gut disorders. Such a functional microbial dysbiosis in IBD context may affect the host–microbiota crosstalk at the mucosal interfaces (eg, shift in microbial associated molecular patterns exposure repertoire, modification of non-commensal clearance activity or trophic alteration of the global ecosystem), favouring the onset or at least the maintenance of digestive tract disorders.

Metagenomic analysis of the human gut microbiota in healthy individuals and patients with inflammatory bowel disease (IBD) highlighted (A) a reduced number of overall non-redundant bacterial genes in patients with IBD. The proportion of individuals having a given number of genes (classes of 100 000 genes were used) is shown. (B) Discrimination of metagenomic composition between healthy individuals (blue) and patients with IBD (red), ulcerative colitis and Crohn's disease, based on an inter-class principal components analysis (adapted from Qin et al 40).
In addition, the worldwide epidemic increase in obesity has boosted research on the impact of diet on host health. The so-called western-style high-fat low-fibre diet dramatically impacts the intestinal microbiota. The composition of the intestinal microbiota of lean and obese people also differs, with an elevated abundance of Firmicutes and a lower abundance of Bacteroidetes in people with obesity that can be altered with diet-induced weightloss.28 Turnbaugh et al have pointed out the existence of a core microbiome between lean and obese individuals while compositional analysis indicated a microbial dysbiosis affecting the Firmicutes/Bacteroidetes ratio.47
Other implications of the composition and functioning of the intestinal microbiota are less intuitive as they seem to trespass physiological boundaries. In humans, the role of the intestinal microbiota in autistic spectrum disorders, for example, remains speculative. Many autistic children are also reported to have gastrointestinal problems,59 which again raises the question of a causal link between events or simply co-occurring events. However, when assessing the effects of intestinal dysbiosis, the gut–brain axis and its bidirectional nature should no longer be ignored.60 ,61 Several animal studies recently challenged the concept of free will and demonstrated that behaviour (anxiety, mating preference, eating behaviour etc) can be modified by intestinal microbiota modulation.62–64
The potential clinical importance of enterotypes distribution and stratification is not to be underestimated: currently, multiple clinical studies are investigating associations between enterotypes and various pathologies such as obesity, diabetes and IBD. If clear links are found, this will open the way to enterotype-based diagnostic and prognostic tests. In addition, enterotypes might be associated with different responses to treatment and even differences in drug metabolism, which may require enterotype-based patient stratification strategies in care management and treatment of various illnesses.
Towards an integrated microbiomic vision of the human body
Metagenomics is a very powerful tool allowing the description of the genetic potential of the microorganisms present in a defined environment, but does not permit monitoring of their activity or gene expression. After assessing the functional capacity of the intestinal microbiota, the next level is to assess the actual functions exerted by the microbiota (figure 3). One way to do so is by analysing the mRNA sequences from the microbiota, the metatranscriptome. Frias-Lopez et al have applied metagenomics and metatranscriptomics to analyse a marine ecosystem.65 They reported that microbial presence and activity are not linked as the most expressed genes were not the most represented ones within the metagenome. They concluded that understanding the fine structure, dynamics and overall impact of a microbial ecosystem requires the combination of these two approaches. Metatranscriptomics analysis of faecal microbiomes from 10 healthy humans66 showed that carbohydrate metabolism, energy production and synthesis of cellular components were the main expressed functionalities. In contrast, housekeeping activities such as amino acid and lipid metabolism were lowered in the metatranscriptome. However, bacterial DNA/RNA extraction methods have an important influence on the downstream results.67 The specific impact of different sampling and nucleic acid extraction methods on metagenomics, and also meta-omics data is currently assessed within the IHMS European funded project International Human Microbiome Standards (IHMS; http://www.microbiome-standards.org/). The main goal of the IHMS is to harmonise practices and facilitate data comparison among projects worldwide.

Integrated meta-omics. The different levels of analyses are represented from phylogeny to metabolomics.
Because mRNA stability is low, microbial gene expression is better captured if sampled physically close to the expression site. However, to compare the transcriptome of bacteria at different intestinal sites (luminal, adherent, mucosal), a protocol uniformly efficient on different types of material (faeces, biopsies, mucus) is mandatory and to our knowledge no satisfactory protocol for bacterial RNA extraction from each of these different matrices has been published so far.
Knowing exactly which proteins and metabolites are active will broaden our knowledge of the intestinal ecosystem. Future challenges for a better understanding of human biology would be clearly related to an integrative perception of our gut microbiome obtained from meta-omics application. In a metaproteomic study of two healthy individuals,68 comparison with metagenomic data drew an interesting picture showing an overabundance of proteins related to post-translational modifications, protein folding and turnover. In contrast, levels of proteins involved in inorganic ion metabolism, cell wall and membrane biogenesis, cell division and secondary metabolite biosynthesis were lower. The data integration process of metagenomic and metaproteomic outcomes has already provided new insights into the genetic capabilities and usage of the human gut microbiome. Meta-metabolomics is currently the most widely used meta-omics approach to study the human gut microbiome. Mainly applied to faecal water extracts, metabolomics has already been used to study numerous digestive tract disorders, including colorectal cancer, IBD and IBS. Described as a suitable approach to be combined with metagenomics,69 it aims to assess the metabolite catalogues of a biome according to a given physiological and environmental context. Its application to healthy and IBD faecal extracts has shed light on metabolome differences among healthy individuals, patients with CD and patients with ulcerative colitis (UC).70 Consistent with previous phylogenetic studies, the IBD metabolome was characterised by reduced levels of butyrate, acetate, and also methylamine and trimethylamine, and an elevated quantity of amino acids. The authors concluded that this was reminiscent of an IBD microbiome composition shift or dysbiosis affecting both short-chain fatty acid production and nutrient absorption. More specifically, the CD metabolome was associated with more extensive inflammation compared with the UC metabolome. Recently, metabolomics was also applied to highlight biomarker metabolites of the CD microbiome.71 Metabolites issued from the metabolism of amino acids, fatty acids especially arachidonic acids, and bile acids were the most notably affected in CD. Another study, including patients with UC and IBS, described an increase in taurine and cadaverine quantities in UC while patients with IBS had higher bile acid concentration and lower levels of branched chain fatty acids compared with relative controls.72 However, changes in short-chain fatty acid and amino acid concentrations were not significant. Finally, metabolomes of patients with colorectal cancer were also investigated and quantities of short-chain fatty acids (butyrate and acetate) and proline/cysteine were respectively lower or higher compared with healthy metabolomes.
The forgotten microorganisms of intestinal microbiota
In almost all investigated ecosystems, there are around 10 phages for every microbial cell, making phages the most abundant biological entities on the planet. Human gut metagenomics highlighted high rates of prophages (ie, bacteriophage genes inserted in the bacterial chromosomes) within bacterial metagenomes. By killing microorganisms, phages greatly influence global biogeochemical cycles, and have been predicted to help maintain microbial species diversity. In 2003, using metagenomics, Breitbart et al described over 1300 viral genotypes within one human faecal sample, most of them corresponding to unknown bacteriophages.73 In addition, Reyes et al demonstrated that faecal viromes, studied in twins and their mothers, were unique to individuals regardless of their degree of genetic relatedness.74 Very recently, Minot et al confirmed these data and reported that viromes were highly sensitive to diet changes.75
More strikingly, much higher amounts of bacteriophages were described in the mucosa of healthy individuals and patients with CD.76 Virus-like particles were counted up to 4×109 per biopsy, and Siphoviridae, Myoviridae and Podoviridae families' morphotypes were dominant. In the context of CD, the ulcerated mucosa exhibited markedly less free bacteriophages than the non-ulcerated mucosa. How the bacteria/phages ratio compares in these very distinct regions of the CD mucosa is a critical question. The current working paradigm for microbial–viral community dynamics is the Lotka Voltera model. Colloquially known as Kill-the-Winner, it predicts that viruses will rapidly and drastically reduce the population of the most abundant microbial species, preventing the best microbial competitors from building up a highly dominant biomass. However, Paterson et al recently validated the Red Queen theory.77 Named after Lewis Carroll's character from Through the Looking Glass, in which the Queen tells Alice: ‘It takes all the running you can do to keep in the same place', this theory hypothesises that bacteria and viruses are in a constant evolutionary arms race. Each has to evolve ever-better ways of outwitting the other to avoid losing out. In addition, these important biomasses of bacteria and phages at the mucosal side are in the vicinity of the toll-like receptors (TLRs) and the immune system. A role for bacteriophages in IBD pathogenesis has been suspected for years and several mechanisms have been hypothesised.78 Upon environmental changes, phages can notably quickly go from a lysogenic into a lytic phase and kill bacteria, which could, in turn, locally decrease specific bacterial populations herein dynamically changing the eubiosis or dysbiosis. Phages might be involved in IBD pathogenesis by changing the recognition patterns of bacteria. By covering bacterial cells, they would change the detection of microbial motifs by TLRs. Finally, phages could be directly recognised by the host and trigger specific modulations. Viruses are known to be recognised by TLR379and TLR13,80 but so far no TLR recognising specific phage components has been described.
Interestingly, the first bacteriophages were isolated from stools of patients recovering from dysenteric diseases.81 D'Herelle rapidly demonstrated that bacteriophages could be used to treat enteric bacterial infections, creating a new field called ‘phage therapy’.82 ,83 Even though their usage in medicine has been controversial, the use of bacteriophages in medicine in east Europe was further developed after the Second World War.84 ,85 Oral administration to humans was shown to be safe86 and nowadays a renewed interest in phage therapy is growing. The first challenge in applying phage therapy in humans is the knowledge of phage host. The specificity of phages is also critical because they could be used as cocktails for larger spectra. Furthermore, the dosage, lethality or survival of phages, isolation and preparation for commercial phage therapy need to be carefully characterised.87
The human gut microbiome: unsolved problems, new directions and emerging challenges
The potential to develop new microbial diagnostic markers of the human metabolic status, to provide early disease diagnostic techniques and new therapeutic strategies, or to maximise the contribution of our microorganisms or probiotics has spawned many human microbiome projects. However, until now, functions encoded by numerous microbial genes found predominant in healthy individuals while missing in patients with disease have so far not been identified. The ongoing collaboration within the IHMC allows metagenomic projects to be supported by strong datasets originating from ongoing genomes sequencing through human microbiome projects. These new experimental and computational tools will facilitate the discovery of impaired pathways to be specifically targeted at larger cohorts using omics technologies.
Biological validation on large human cohorts is required to demonstrate the robustness of predictors of diseases. The classification of human gut microbiome into three enterotypes opens up questions about their functional impact on human health and individual ‘classifications’. Even though larger studies analysing more diverse cohorts will be necessary to confirm and possibly refine the enterotype concept, whether the enterotypes correspond to ecosystems with different metabolic properties, but also distinct community–host interactions, is a key question. The enterotypes stability, or ‘enterostates’ variability, also needs to be investigated because it will condition their use in nutritional and clinical trials. The original enterotype work had very few family members; although some of these were in different enterotypes, a (partial) genetic/immunological/maternal effect should not be ruled out. Also, current studies have focused on limited and western-diet biased cohorts. A broader sampling of the human population, using bigger cohorts88 with multiple age groups, wider geographical spread including remote populations, and for longer periods of time will increase our understanding of the true nature of enterotypes and pinpoint their underlying origins.
If faecal sample analysis as a surrogate for the entire gut microbiota is necessary for large cohort screening and specific biomarker detection, determining the mechanisms will require the meta-omics description of the mucosal microbiota. Recently, Wang et al showed that, from the metagenome associated with the right and left colonic mucosa,89 functional diversity was comparable to that reported in faeces, with an overall functionality mainly supported by genes associated with carbohydrate, protein and nucleic acid utilisation. At mucosal interfaces, a loss of bacteria–host crosstalk was described in patients with UC by combining a description of the microbiota and host transcriptomic profiles.26 The co-sequencing and direct interfacing of human and microbiome genomes is now feasible and, while computationally challenging, it should uncover a new link between human–microbes co-evolution and pathways disturbance in disease conditions.
More and more evidence from animal studies shows a genetic effect on microbiome composition (especially with regard to innate immunity-related mutations).90–92 Interestingly, unaffected relatives of patients with IBD also show subclinical microbial dysbiosis.26 ,93 ,94 However, the genetic and environmental risk factors they share with their diseased relatives seem to be counterbalanced. For example, unaffected relatives of patients with IBD have more butyrate-producing bacteria. Therefore, metagenomic analysis of unaffected relatives will be important to answer the recurrent question of whether the dysbosis is the ‘cause or consequence’ of the disease.
Gnotobiotic animal models, colonised by synthetic enterotypes microbiota, could represent suitable tools for translational research to characterise the impact of genetic defects, diet alteration, probiotic intake and microbiome transplant, and to provide the proof of principle needed to direct and interpret human studies.
Finally, metagenomics has strongly increased our capacity to mine the clinical and biological data that will soon allow construction and experimental validation of biological models. Mathematical models of the human gut, first focusing on host–microbiota interaction or on specific microbial pathways, should soon be integrated in a global model framework capturing key inter-relationships between host factors (genetic, epigenetic, immunological and serological data) and its microbiome (figure 4). Since many diseases are not spatially constrained to one organ, as discussed at the October 2005 IHMC foundation meeting, the understanding of human–microbiota interactions requires the analysis of all human colonised body site metagenomes. This will lead to the development of novel tools for preclinical prognostics and early diagnostics; definition and validation of biological networks relevant to different disease pathophysiologies; and identification of new therapeutic targets and intervention/prevention strategies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
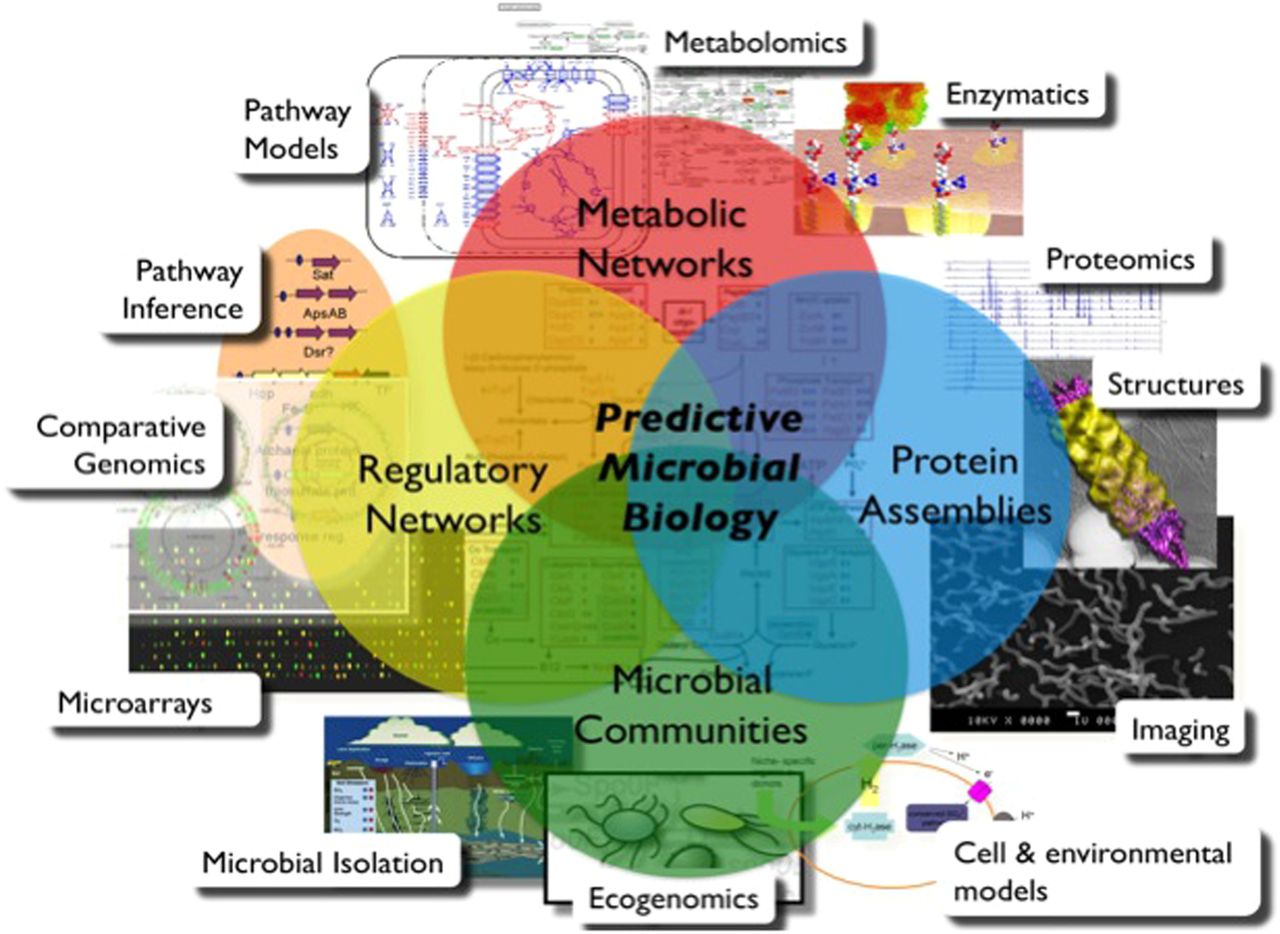
An integrative approach to study microbial systems. Modified from http://ms.lbl.gov/, courtesy of T Northen. The comprehension of a complex microbial system and the prediction of its dynamic require the integration of heterogeneous data obtained using several of the 12 methods depicted in the rectangles. For example, microbial isolation and metagenomics allow characterization of the microbial communities, and they can now be combined with metabolomics and pathway models to obtain the metabolic network (of the organisms or) of the whole community. In addition, regulation of these microbial systems, critical in health and disease, can be defined using in silico comparative genomics, to infer pathways that might be further validated against microarray data.
Glossary
Core human microbiome
Whatever is shared in a given body habitat among all or the vast majority of human microbiomes. A core microbiome may include a common set of organisms (phylotypes or their genomes), gene or protein families, and/or metabolic capabilities. Microbial genes that are variably represented in different humans may contribute to our distinctive metabolic attributes (metabotypes).
Dysbiosis
Alteration of the microbiota in comparison to the normal, healthy state. Dysbiosis refers to a condition of microbial imbalances compared with the ‘eubiosis’ condition, a state of equilibrium between bacterial symbionts and pathobionts. Dysbiosis is mostly reported in the digestive tract or on the skin, but can also occur on any exposed surface or mucous membrane. Dysbiosis can be observed at several analytical levels, from diversity to compositional or functional imbalances.
Meta-omics
Meta-omics includes shotgun sequencing of microbial DNA isolated directly from a given environment; high-throughput screening of expression libraries, constructed from cloned community DNA, to identify specific functions such as antibiotic resistance (functional metagenomics); profiling of RNAs and proteins produced by a microbiome (meta-transcriptomics and meta-proteomics); and identification of a community's metabolic network (metabolomics).
Metabolomics
The characterisation by mass spectroscopy, nuclear magnetic resonance, or other analytical methods of metabolites generated by one or more organisms in a given physiological and environmental context.
Metagenomics
The study of metagenomes, genetic material recovered directly from environmental samples. Metagenomics is the genomic analysis (analysis of all the DNA in an organism) applied to all the microorganisms of a microbial ecosystem without previous identification. Metagenomics is an emerging field encompassing culture-independent studies of the structures and functions of microbial communities and their interactions with the habitats they occupy to understand their biological diversity.
Microbiota
A microbial community, including bacteria, archaea, eukarya, and viruses, which occupies a given habitat.
Microbiome
The totality of microbes, their genetic elements (genomes), and environmental interactions in a defined environment. In this sense, the human ‘micro’biome would be defined as the collection of microorganisms associated with the human body, and their collective genomes would constitute a metagenome. However, the term microbiome is now commonly used to refer to the collective genomes present in members of a given microbiota.
Open reading frames
DNA sequence that does not contain a stop codon in a given reading frame. It allows initial identification of candidate protein coding regions in a DNA sequence. A simple gene prediction algorithm for prokaryotes might look for a start codon followed by an open reading frame that is long enough to encode a typical protein, where the codon usage of that region matches the frequency characteristic for the given organism's coding regions.
Phylotype
A phylogenetic group of microbes, currently defined by a threshold per cent identity shared among their small subunit (16S) rRNA genes (eg, ≥97% for a ‘species’ level phylotype).
Web resources
-
IHMC: http://www.human-microbiome.org/. The International Human Microbiome Consortium focuses on generating a shared comprehensive data resource that enables investigators to characterise the relationship between the composition of the human microbiome and human health and disease.
-
MetaHIt: http://www.metahit.eu/. MetaHIT is a project financed by the European Commission under the 7th Framework Program and coordinated by S D Ehrlich, INRA, France. The consortium gathers 13 partners from academia and industry, from a total of eight countries.
-
HMP: http://commonfund.nih.gov/hmp/overview.aspx. The aim of the Human Microbiome Program, funded by the NIH in the USA, is to characterise microbial communities found at multiple human body sites and to look for correlations between changes in the microbiome and human health.
-
Gold: http://www.genomesonline.org/cgi-bin/index.cgi. Genomes Online Database, is a web resource for comprehensive access to information regarding genome and metagenome sequencing projects, and their associated metadata, around the world.
Key messages
-
The human gut contains 1014 bacteria (mass of 2 kg in the gut), 10 times the number of human cells, 150 times more genes than the human genome.
-
Other organisms in the human gut belong to archaea, viruses (prokaryotic and eukaryotic), parasites, fungi.
-
The human gut microbiota plays a key role in human health, gastrointestinal disease and other diseases.
-
Commensal bacteria develop mechanisms of crosstalk with human cells that play key roles in homeostasis maintenance.
-
Metagenomics allows the description of the combined genomes of the microorganisms present in the gut, even non-cultured ones, giving access to their potential functions.
-
Human gut contains an important diversity and high number of non-redundant bacterial genes.
-
Based on gut microbial composition distribution, the human population can be subdivided into enterotypes.
-
A better description of the human gut microbiota will lead to description of biomarkers of disease, development of new probiotics/prebiotics, new therapies such as faecal transplantation.
Acknowledgments
The authors would like to thank Nicolas Lapaque for careful proofreading of the manuscript.
References
Footnotes
-
Funding The authors would like to gratefully acknowledge the financial support of the French National Agency for Research, the 7th European Framework Program (MetaHIT project and Crosstalk project). JR is supported by the Research Foundation Flanders (FWO) and the Agency for Innovation by Science and Technology (IWT).
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.
-
Data sharing statement Available data in this paper will be shared upon request to the corresponding author.