Article Text

Abstract
Objective CHIP (carboxy terminus of Hsc70 interacting protein) is an E3 ubiquitin ligase that can induce ubiquitination and degradation of several tumour related proteins, and acts as a suppressor of tumour metastasis. This study explored the biological function and clinical significance of CHIP in gastric cancer (GC).
Methods The prognostic value of CHIP expression was evaluated using tissue microarray and immunohistochemical staining in two independent human GC cohorts. The role of CHIP on tumorigenicity and angiogenesis was determined in vitro and in vivo.
Results CHIP expression was significantly decreased in GC lesions compared with paired non-cancerous tissues. Low tumoral CHIP expression significantly correlated with clinicopathological characteristics in patients, as well as with shorter overall survival in both cohorts. Multivariate Cox regression analysis revealed that CHIP expression was an independent prognostic factor for human GC patients. Moreover, CHIP overexpression impeded the formation of anchorage independent colonies in soft agar, suppressed the growth of xenografts in nude mice and inhibited endothelial cell growth and tube formation by suppressing nuclear factor κB (NF-κB) mediated interleukin 8 (IL-8) expression in vitro. In vivo studies also confirmed that CHIP inhibited blood vessel formation and recruitment of CD31 positive cells in matrigel plugs. Also, CHIP interacted with NF-κB/p65 and promoted its ubiquitination and degradation by proteasome, terminating NF-κB activity and inhibiting IL-8-induced angiogenesis, which correlated with subsequent tumour metastasis.
Conclusions Decreased CHIP expression in GC resulted in increased angiogenesis and contributed to GC progression and poor prognosis. CHIP expression is a GC candidate clinical prognostic marker and a putative treatment target.
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
CHIP includes a tetratricopeptide repeat domain and a U-box domain, which functions as a link between the chaperone and proteasome systems.
-
CHIP functions as a tumour suppressor because it can induce ubiquitination and degradation of several oncogenic proteins.
-
CHIP inhibits the metastatic potential of human breast cancer, and low CHIP expression is associated with shorter survival of breast cancer patients.
What are the new findings?
-
CHIP expression is significantly decreased in gastric cancer (GC) lesions compared with paired non-cancerous tissues, which may be associated with CHIP promoter methylation in GC cells.
-
Low tumorous CHIP expression significantly correlated with clinicopathological characteristics, as well as with shorter overall survival in GC patients.
-
CHIP was identified as an independent prognostic factor for patients with resectable GC and could add significant prognostic value to the established prognostic factors (eg, TNM stage, histological type and tumour diameter).
-
CHIP interacted with nuclear factor κB (NF-κB)/p65 and promoted its ubiquitination and degradation by proteasome, which terminates NF-κB activity and inhibits interleukin 8 induced angiogenesis that also correlated with subsequent tumour metastasis.
How might it impact on clinical practice in the foreseeable future?
-
CHIP may serve as a promising prognostic marker for GC, and restoration of CHIP may be a novel strategy for antiangiogenic therapy for human GC.
Introduction
Despite the improved prognosis of patients with gastric cancer (GC) resulting from early diagnosis, radical surgery and the development of adjuvant therapy, GC remains the fourth most common cancer in the world and the second leading cause of cancer related death worldwide.1 The high mortality rates in GC patients are associated with metastatic disease.2 ,3 Five year survival is fairly high for patients with localised disease (62%) but dramatically decreased when the tumour has already spread to regional lymph nodes (22%) or distant organ sites (3%).4
Metastasis is the result of several sequential steps of tumour development.5 Angiogenesis is a critical hallmark of malignancy and is induced surprisingly early during the multistage development of invasive cancers both in animal models and in humans.6 Although angiogenesis is an important aspect of tumour growth and metastasis and considered as an important predictor of overall survival in GC,7 little is known about the molecular events critical to GC angiogenesis. Unravelling the factors driving this process is thus important for future therapeutic interventions also.
The nuclear factor κB (NF-κB) is a critical transcription factor in various cancers that regulates genes associated with a variety of cellular functions, such as cell survival, proliferation, angiogenesis and cancer metastasis.8–11 However, excessive and prolonged activation of NF-κB can result in human diseases, such as inflammation and carcinogenesis.10 ,12 Thus NF-κB activation must be terminated at the appropriate time for maintaining normal function. p65 (also called RelA) is a member of the NF-κB family, which is normally sequestered in the cytoplasm of non-stimulated cells by inhibitors of NF-κB (IκBs). Once cells are exposed to extracellular stimuli, IκBs become phosphorylated, ubiquitinated and degraded by the proteasomes, leading to p65 activation.13 Recently, a novel pathway of terminating NF-κB activation was revealed showing that nuclear p65 protein level is regulated by ubiquitin–proteasome dependent degradation.14–18 Therefore, the molecules involved in controlling the aberrant activation of NF-κB signalling are attractive targets for therapeutic strategies against cancer.
CHIP (carboxy terminus of Hsc70 interacting protein), encoded by the STUB1 gene, includes a tetratricopeptide repeat (TPR) domain at its amino terminus interacting with the molecular chaperones Hsc70-Hsp70 and Hsp90 protein, and a U-box domain at its carboxy terminus with E3 ubiquitin ligase activity, which functions as a link between the chaperone and proteasome systems.19 ,20 Substantial evidence indicates that CHIP functions as a tumour suppressor because it induced ubiquitination and degradation of several oncogenic proteins, such as mutant p53,21 oestrogen receptor α,22 c-ErbB2/neu,23 Dbl,24 Smad3,25 hypoxia inducible factor 1α,26 Runx1,27 Met receptor28 and SRC-3.29 A recent study showed that CHIP overexpression inhibits metastatic potential and knockdown of CHIP increased the microvessel density in breast tumours.29 However, the precise mechanisms of CHIP in angiogenesis have not yet been identified. It was also shown that CHIP expression was significantly lower in breast cancers compared with normal mammary glands,29 ,30 which also correlated with shorter survival of breast cancer patients.30 However, the possible function of CHIP in the development and progression of GC and the prognostic role in GC patients remains unclear. In this study, the biological function and clinical significance of CHIP in GC was examined.
Materials and methods
Patients and specimens
Patients were recruited from the Nantong Cancer Hospital, Nantong City, Jiangsu Province, China. Two independent GC cohorts from two time periods were studied. The training cohort included 83 patients who underwent radical gastrectomy from 1 May 1990 to 1 June 1995 and the validation cohort consisted of all 640 surgical cases from 1 December 2000 to 1 April 2005 (for details, see the methods section of the supplementary data; supplementary data are available online only).
Tissue microarray construction and immunohistochemistry
GC tissue microarrays (TMAs) were created by a contract service at the National Engineering Centre for Biochip, Shanghai, China and a standard protocol was used for immunostaining of the TMAs, as described in the supplementary data (supplementary data are available online only).
Assessment of immunohistochemistry
Staining of CHIP in tissue was scored independently by two pathologists blinded to the clinical data, by applying a semiquantitative immunoreactivity score (IRS) in the training cohort, as reported elsewhere,31 and the optimum cut-off value of IRS was obtained by receiver operating curve (ROC) analysis, as described in the supplementary data (the supplementary data are available online only). Under these conditions, samples with IRS 0–4 and IRS 6–12 were classified as low and high expression of CHIP, respectively. After establishing the immunohistochemical assessment criteria in the training cohort, expression of CHIP in the validation cohort was scored using the same procedure. Blood vessels were assessed by staining endothelial cells for CD31, as described in the supplementary data (supplementary data are available online only).
Animals, cell lines and reagents
Animals, cell lines and reagents used are described in the supplementary data (supplementary data are available online only).
Plasmids and stable or transient transfections
CHIP overexpression and knockdown GC cells were produced by stable or transient transfection methods as GC cells are likely to have deregulated CHIP levels. The constructs are described in the supplementary data (supplementary data are available online only). The plasmids were transiently transfected into cells using Lipofectamine 2000 (Invitrogen, Shanghai, China) following the manufacturer's protocol. The cells from a single clone were stably selected with G418 at a final concentration of 600 μg/ml.
Small interfering RNA
Small interfering RNA (siRNA) specific for CHIP and non-specific control siRNA was synthesised (Ribobio, Guangzhou, China) and transfected using Lipofectamine 2000. The sequences of siRNA are described in the supplementary data (supplementary data are available online only). p65 siRNA was obtained from Cell Signaling Technology (Beverly, Massachusetts, USA).
Quantitative real time RT-PCR assay
Quantitative real time PCR was carried out in triplicate with SYBR Green PCR Master Mix using a 7900HT qPCR system thermal cycler (Applied Biosystems, Foster City, California, USA) and performed as described previously.32 The primers and conditions are described in the supplementary data (supplementary data are available online only).
DNA extraction and methylation specific PCR
Genomic DNA was extracted from gastric tissues using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). DNA (1 μg) was treated with sodium bisulfite using EpiTect Bisulfite Kits (Qiagen) and then the modified DNA was amplified by methylation specific PCR, as described in the supplementary data (supplementary data are available online only).
Subcellular fractionation, western blotting and ELISA analysis
Nuclear extracts were obtained using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, Illinois, USA). Western blots were performed as previously reported.32 Antibodies are described in the supplementary data (supplementary data are available online only).
Secreted interleukin (IL)-8 protein in the conditioned medium was measured by a human IL-8 ELISA kit from R&D Systems (Minneapolis, Minnesota, USA) according to the manufacturer's instructions.
FLAG affinity purifications and co-immunoprecipitation
FLAG affinity purifications and co-immunoprecipitation were carried out for both endogenous and ectopically expressed proteins using the routine methods described in the supplementary data (supplementary data are available online only).
Indirect immunofluorescent microscopy
Indirect immunofluorescent microscopy, performed as described previously,33 is briefly outlined in the supplementary data (supplementary data are available online only).
Adhesion, invasion and cell viability assay
Adhesion and invasion assays were performed as described previously,33 and briefly outlined in the supplementary data (supplementary data are available online only).
Human umbilical vein endothelial cell growth and tube formation assay
GC cells were cultured in 60 mm plates with fresh complete medium for 24 h, and 2 ml of conditioned medium were collected. The detailed procedures of the human umbilical vein endothelial cell (HUVEC) growth and tube formation assay are described in the supplementary data (supplementary data are available online only).
Luciferase reporter gene assay
Luciferase activity was measured with a dual Luciferase Reporter Assay System (Promega, Madison, Wisconsin, USA) and the detailed procedures are described in the supplementary data (supplementary data are available online only).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays were performed with a Biotin Gel Shift Kit (Pierce, Rockford, Illinois, USA), as described previously,32 and briefly outlined in the supplementary data (supplementary data are available online only).
Tumour xenograft, angiogenesis assay in vivo and CD31 immunofluorescent staining
Tumour xenograft and angiogenesis assay in vivo are described in the supplementary data (supplementary data are available online only). Immunofluorescent staining for CD31 expression was then performed using antimouse CD31 FITC labelled antibody (eBioscience, San Diego, California, USA). Sections were counterstained with DAPI. The number of CD31 positive cells was counted in five random fields for each group.
Statistical analysis
The associations between CHIP expression and clinicopathological parameters were evaluated by Fisher's exact test. Differences in IRS for CHIP staining in primary tumours and their corresponding non-tumours were assessed by the Wilcoxon test (grouped). Probability of differences in overall survival (OS) was ascertained by the Kaplan–Meier method, with a log rank test for significance. Univariate or multivariate Cox regression analysis was used to estimate the HRs and their 95% CI. Then we analysed the predictive value of the parameters using time dependent ROC curve analysis for censored data and calculated the AUC of the ROC curves.34 We evaluated the performances of different scores by plotting (t, AUC [t]) for different values of follow-up time (t). A p value of <0.05 was deemed statistically significant. All statistical analyses were performed by Statistical Analysis System software (V.9.1.3; SAS Institute), STATA statistical software (V.10.1; Stata Corp) and R software (V.2.10.1; The R Foundation for Statistical Computing).
Results
Reduced CHIP expression in gastric cancer versus non-cancer tissues
mRNA and protein levels of CHIP were found to be significantly lower in 12 of 16 (75%) and in 14 of 16 (87.5%) gastric tumours compared with the paired normal gastric mucosa by real time PCR and western blot, respectively (figure 1A,B). Immunohistochemical staining of the gastric tissues showed that CHIP seemed to be predominantly localised in the cytoplasm of the GC cells but presented in both the nucleus and cytoplasm of normal cells. Moreover, expression of CHIP was significantly decreased in GC cells (figure 1C). These results were confirmed in TMA of GC patients which showed that the expression of CHIP was significantly decreased in 64 of 74 (86.5%) of GCs compared with matched normal gastric tissues (p<0.001; figure 1D). We also found tumours from diffuse-type patients with decreased levels of CHIP mRNA compared with those from intestinal-type tissues (p=0.035; figure 1E). Additionally, methylation specific PCR analysis detected CHIP promoter methylation in all GC tissues and in none of their corresponding non-cancerous tissues, and unmethylated CHIP promoter in all normal tissues and in two of 12 (16.7%) cancer tissues (figure 1F).
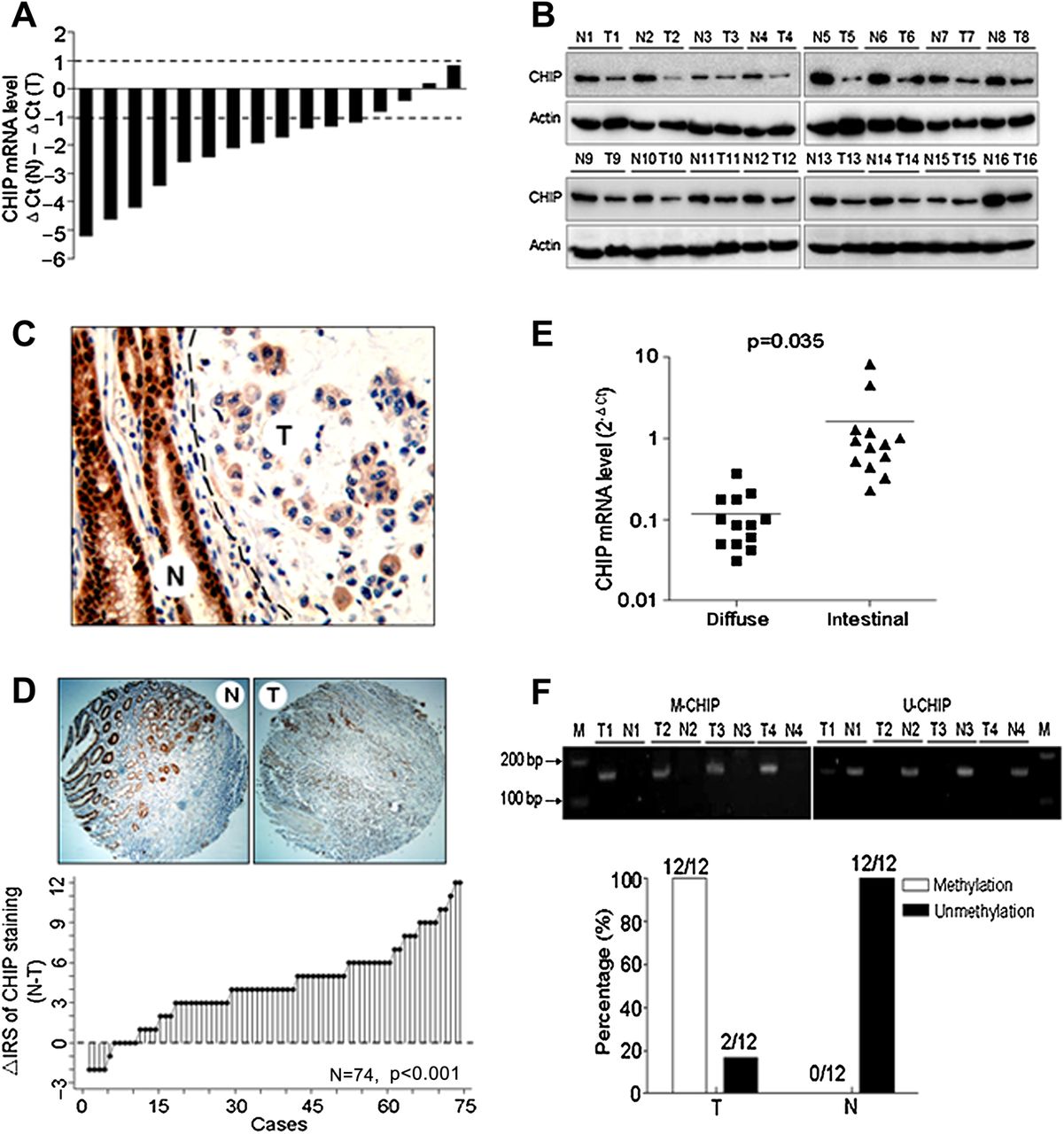
Expression of CHIP (carboxy terminus of Hsc70 interacting protein) is decreased in human gastric cancer (GC). (A) Determination of CHIP mRNA level in 16 cancer tissues and paired non-cancerous normal tissues of GC patients by real time PCR. ΔCt(N), Ct value of GAPDH was subtracted from the Ct value of CHIP of gastric normal tissue. ΔCt(T), Ct value of GAPDH was subtracted from that of CHIP of the paired cancer tissue tumour. Bar value (ΔCt(N) − ΔCt(T)) represents the difference in CHIP mRNA between normal tissue and paired tumour. Bar value =−1 indicates that CHIP mRNA of tumour is 2−1-fold of that of paired normal tissue. Bar value =1 indicates that CHIP mRNA of tumour is 21-fold of that of paired normal tissue. Bar value ≤−1 indicates that the expression of CHIP is decreased in tumours. Bar value ≥1 indicates that the expression of CHIP is increased in tumours. (B) CHIP protein level in patients as in (A) was analysed by western blotting. The level of each protein was normalised against β actin. (C) Representative images of immunohistochemical staining of tissues with CHIP antibody. Normal (N) or tumour (T) tissues are marked with dotted lines; original magnification, ×200. (D) Top: immunohistochemical staining of tissue microarray with CHIP antibody; original magnification, ×100. Bottom: the distribution of the difference in CHIP staining (Δ IRS=IRS N-IRS T). Immunoreactivity score (IRS) of CHIP staining was available from 74 pairs of tissues; p values were calculated with the Wilcoxon test. (E) Relationship between histological type and CHIP mRNA expression in 26 GC tissues. Total RNA was prepared from 13 diffuse-type or 13 intestinal-type tumour tissues and the CHIP mRNA level was quantified using real-time PCR. (F) DNA methylation analysis of CHIP by methylation specific PCR in 12 GC and paired normal gastric tissues. M, DNA marker; M-CHIP, methylated CHIP products; N, paired non-cancerous gastric tissues; T, gastric cancerous tissues; U-CHIP, unmethylated CHIP products.
Decreased CHIP expression correlates with clinicopathological characteristics and a shorter survival in gastric cancer patients
As shown in table 1, protein expression of CHIP in the cancerous tissues of the training cohort significantly correlated with clinicopathological features, such as depth of invasion, lymph node metastasis, distant metastasis, TNM stage and histological type (p<0.05 for all). Moreover, these findings, except for distant metastasis (p=0.383), were confirmed in the validation cohort of GC patients.
Relationship between expression levels of CHIP and demographic and clinicopathological features of the individuals in two cohorts of gastric cancers
In the training cohort, Kaplan–Meier analysis revealed that low CHIP staining was significantly correlated with a poorer overall 5 year survival of all GC patients (p<0.001, log rank test; figure 2A). The median survival time of patients whose primary GC scored high for CHIP expression was more than 60 months whereas low CHIP expression correlated with a shortened median survival time of 23 months. This finding was confirmed in the validation cohort (p<0.001, figure 2B). Next, multivariate Cox regression analysis indicated that CHIP expression was an independent positive prognostic factor in both cohorts (HR=0.34, 95% CI 0.15 to 0.76, training cohort; HR=0.21, 95% CI 0.16 to 0.26, validation cohort; table 2).

Kaplan–Meier curves depicting overall survival according to expression patterns of CHIP (carboxy terminus of Hsc70 interacting protein) in the training cohort (n=77) (A) and in the validation cohort (n=493) (B). The p values were calculated using the log rank test. Time dependent receiver operating curve (ROC) analyses (C–D). Figures show the time dependent ROC analysis for the clinical risk score (TNM stage, histological type and tumour diameter), CHIP risk score and the combined CHIP and clinical risk score in the training cohort (C) and in the validation cohort (D). AUC, area under the curve.
Multivariate Cox regression analysis of CHIP and clinical variables predicting survival in two cohorts of gastric cancers
To further evaluate the predictive ability of CHIP expression, we conducted a time dependent ROC analysis which indicated that the combination of clinical risk score (TNM stage, histological type and tumour diameter) and CHIP risk score contributed much more than either one alone in both cohorts. The AUC at year 5 was 0.688 (95% CI 0.550 to 0.826) and 0.723 (95% CI 0.675 to 0.770) for a clinical risk score, whereas it was significantly increased to 0.760 (95% CI 0.621 to 0.899) and 0.842 (95% CI 0.806 to 0.878) for the combination of the clinical risk score with the CHIP risk score in the training cohort (figure 2C) and in the validation cohort (figure 2D).
CHIP suppresses gastric cancer cell growth and angiogenesis
Investigation of CHIP regulation of GC cell growth indicated that cell proliferation rates were similar between CHIP overexpressed (CHIPOE) or CHIP siRNA cells and corresponding controls under normal culture conditions (figure 3A). However, in the soft agar colony formation assay there were 50% fewer colonies formed in CHIPOE stable cells and, in agreement with this, knockdown of CHIP expression significantly increased the number of colonies compared with control cells (figure 3B). To further investigate the role of CHIP in tumorigenesis in vivo, stable CHIPOE and vector control BGC823 cells were injected subcutaneously into nude mice and tumour growth was monitored. The data showed that tumour growth of BGC823 cells was suppressed in the CHIP overexpression group (figure 3C,D).

CHIP (carboxy terminus of Hsc70 interacting protein) suppresses gastric cancer (GC) tumorigenesis and angiogenesis. (A) CHIP overexpressed (CHIPOE), CHIP small interfering RNA (siRNA) and their corresponding control (ctrl) cells were grown in 96 well plates for 12, 24, 36 or 48 h. Cell survival was determined by the MTT assay. Results were averaged from sextuplet samples with SD shown. (B) CHIP inhibits anchorage independent growth in BGC823 cells. Cells were plated in soft agar dishes. After incubation for 3 weeks, colonies were photographed as shown in the left panel (magnification: ×40) and were counted under a light microscope as shown in the right panel. Results are means ± SD from three independent experiments. (C) CHIP overexpression inhibits the growth of GC xenografts in nude mice. The stably selected CHIPOE and vector BGC823 cells were subcutaneously injected into nude mice. Thirty-eight days after the injections, the mice were killed and tumour tissues were collected. Upper panel shows the photographs of the tumours. The lower panel shows tumour growth curves in nude mice inoculated with vector or CHIPOE cells (left) and the weight of the tumours after 38 days (right). Data are presented as mean ± SEM (n=5). (D) CHIP protein levels in the tumour tissues were analysed by immunoblotting. (E) Sections of tumours from injected nude mice were stained with CD31 and CHIP antibodies by immunohistochemistry. (F–G) Expression of CHIP in BGC823 cells negatively regulates human umbilical vein endothelial cell (HUVEC) growth and tube formation. (F) CHIP overexpression in BGC823 cells inhibits HUVEC growth whereas CHIP knockdown promotes HUVEC growth. Data are presented as means ± SD from three independent experiments. (G) Representative images in the left panel show that CHIP overexpression in BGC823 cells inhibits HUVEC tube formation whereas CHIP knockdown promotes HUVEC tube formation. In the right panel, numbers of tubes formed per field were counted in five random fields for CHIPOE, CHIP siRNA and corresponding control groups, respectively. *p<0.05, **p<0.01.
Microvessel density evaluated by CD31 immunohistochemical staining showed a significant reduction in tumour tissues of the CHIPOE group compared with vector controls (figure 3E). To further study the functional role of CHIP in GC angiogenesis, HUVEC growth and tube formation in vitro were investigated. The growth of HUVECs in conditioned medium from CHIPOE BGC823 cells was inhibited by 48% and, in line with this, CHIP knockdown promoted HUVEC growth by 1.82-fold compared with the corresponding controls (figure 3F). The average number of complete tubular structures formed by HUVECs was significantly decreased in conditioned medium from CHIPOE BGC823 cells but increased in that from CHIP siRNA cells compared with corresponding controls (figure 3G).
CHIP inhibits NF-κB activity and expression of its target proangiogenesis genes
We then investigated whether CHIP exerts its inhibitory effect on GC cells angiogenesis through inhibiting NF-κB activity. It was shown that NF-κB activity was increased following cotransfection with NF-κB–p65 expression plasmid or exposure to NF-κB–p65 activator (either tumour necrosis factor α (TNFα) or lipopolysaccharide) whereas it was significantly reduced with or without these treatments in CHIP overexpressing cells (figure 4A). In contrast, knockdown CHIP enhanced NF-κB activity. This effect was further abolished by knockdown of NF-κB–p65 expression with a specific siRNA or suppression of NF-κB–p65 activity with a specific inhibitor (Bay-11) (figure 4B).

CHIP (carboxy terminus of Hsc70 interacting protein) inhibits nuclear factor κB (NF-κB) activity and its target proangiogenesis gene expression, which suppresses interleukin (IL)-8 dependent angiogenic potential of endothelial cells in vitro. (A–B) Overexpression of CHIP inhibits but knockdown CHIP enhances NF-κB activity. BGC823 cells were cotransfected with the recombinant, including either myc-CHIP or vector control with or without Flag-p65 (A), or CHIP small interfering RNA (siRNA) or control (ctrl) siRNA with or without p65 siRNA (B), together with the NF-κB reporter plasmids. After 24 h, the transfected cells were cultured in the presence or absence of 20 ng/ml of tumour necrosis factor α (TNFα) for 24 h or 1 μg/ml of lipopolysaccharide (LPS) for 8 h (A), or 5 μM of Bay 11 for 1 h (B), and then the reporter activity was examined. The means ± SD of triplicate experiments are shown. (C) CHIP alters the DNA affinity of NF-κB–p65. Electrophoretic mobility shift assay was performed using nuclear protein extracts from vector, CHIP overexpressed (CHIPOE), ctrl siRNA or CHIP siRNA BGC823 cells with or without treatments with 20 ng/ml of TNFα for 30 min or 5 μM of Bay 11 for 1 h and the oligonucleotide probes bearing NF-κB consensus binding sequence. A major shifted band is indicated by an arrow. Lane 10 represents competition analysis using 100-fold unlabelled probes. (D) BGC823 cells were transiently transfected with vector control or myc-CHIP plasmid. After 48 h, the transfected cells were treated with or without 20 ng/ml of TNFα for 30 min and then fixed, and immunostained with anti-myc or anti-p65 antibody, as indicated by the single arrows; the double arrows denote that the myc-CHIP was absent in the cell due to ineffective transfection. Representative images were photographed and coloured using a Zeiss LSM 510 confocal microscope system. (E) CHIP suppresses NF-κB targeting proangiogenesis gene expression. BGC823 cells were transiently transfected with vector or myc-CHIP plasmid for 48 h and ctrl siRNA or CHIP siRNA for 72 h, respectively. CHIP, IL-8, IL-6, matrix metalloproteinase (MMP)-2 and vascular endothelial growth factor (VEGF) mRNA in the cells were determined with real time RT-PCR. (F–G) NF-κB is required for CHIP regulated expression of IL-8 in gastric cancer cells. BGC823 cells were transfected with myc-CHIP or vector, together with Flag-p65 plasmid (F), either CHIP siRNA or ctrl siRNA, together with p65 siRNA (G), then IL-8 mRNA levels and protein levels in the conditioned media in the cells were determined by real time RT-PCR analysis (left panel) and ELISA (right panel), respectively. (H–I) Addition of IL-8 recombinant protein (0.4 ng/ml) to the conditioned medium rescued CHIP suppressed endothelial cell tube formation (H) while application of IL-8 antibody (400 ng/ml) to neutralise the IL-8 in conditioned medium abrogated CHIP knockdown enhanced tube formation (I). *p<0.05, **p<0.01.
Electrophoretic mobility shift assay indicated that NF-κB affinity was decreased in CHIP overexpressing cells with or without TNFα treatment (figure 4C, lane 7 and 9), and increased in CHIP knockdown cells (lane 3) compared with corresponding controls. Furthermore, we performed immunofluorescence to examine the effects of CHIP on p65 localisation (figure 4D). It was shown that myc-CHIP inhibited p65 translocation into the nucleus and attenuated p65 protein levels with or without TNFα treatment.
Moreover, our data revealed that CHIP overexpression decreased NF-κB responsive genes, such as IL-8, matrix metalloproteinase (MMP)-2 and vascular endothelial growth factor (VEGF) mRNA expression by 52%, 24% and 20%, respectively; and CHIP knockdown elevated these mRNA levels by 2.0-, 1.4- and 1.3-fold, respectively, compared with corresponding controls (figure 4E). However, IL-6 mRNA expression was not affected by CHIP alteration. These results were confirmed in the xenografts in nude mice which showed that p65 and IL-8 expression were reduced in CHIP overexpressing tissues (supplementary figure S3A–C; supplementary figure S3 is available online only).
ELISA was used to confirm alterations of secreted IL-8 protein corresponding to CHIP modulation. We found that CHIP overexpression decreased IL-8 protein in the medium from 0.85 ng/ml to 0.52 ng/ml (figure 4F). In CHIP knockdown cells, IL-8 protein increased from 0.90 ng/ml to 1.75 ng/ml in the medium (figure 4G). CHIP overexpression also decreased p65 induced IL-8 mRNA and protein production (figure 4F) whereas silencing of p65 blocked CHIP siRNA induced IL-8 mRNA and protein production (figure 4G).
To confirm the role of IL-8 in CHIP regulated GC cell angiogenesis, we performed IL-8 rescue and IL-8 blocking assays. We found that the inhibited tubular structure formations in the conditioned medium from CHIP overexpressing BGC823 cells can be rescued by addition of 0.4 ng/ml of recombinant IL-8 (figure 4H). Application of sufficient IL-8 antibody abrogated the elevated tube formation in that from CHIP knockdown cells (figure 4I).
CHIP promotes NF-κB ubiquitination for degradation by the proteasome
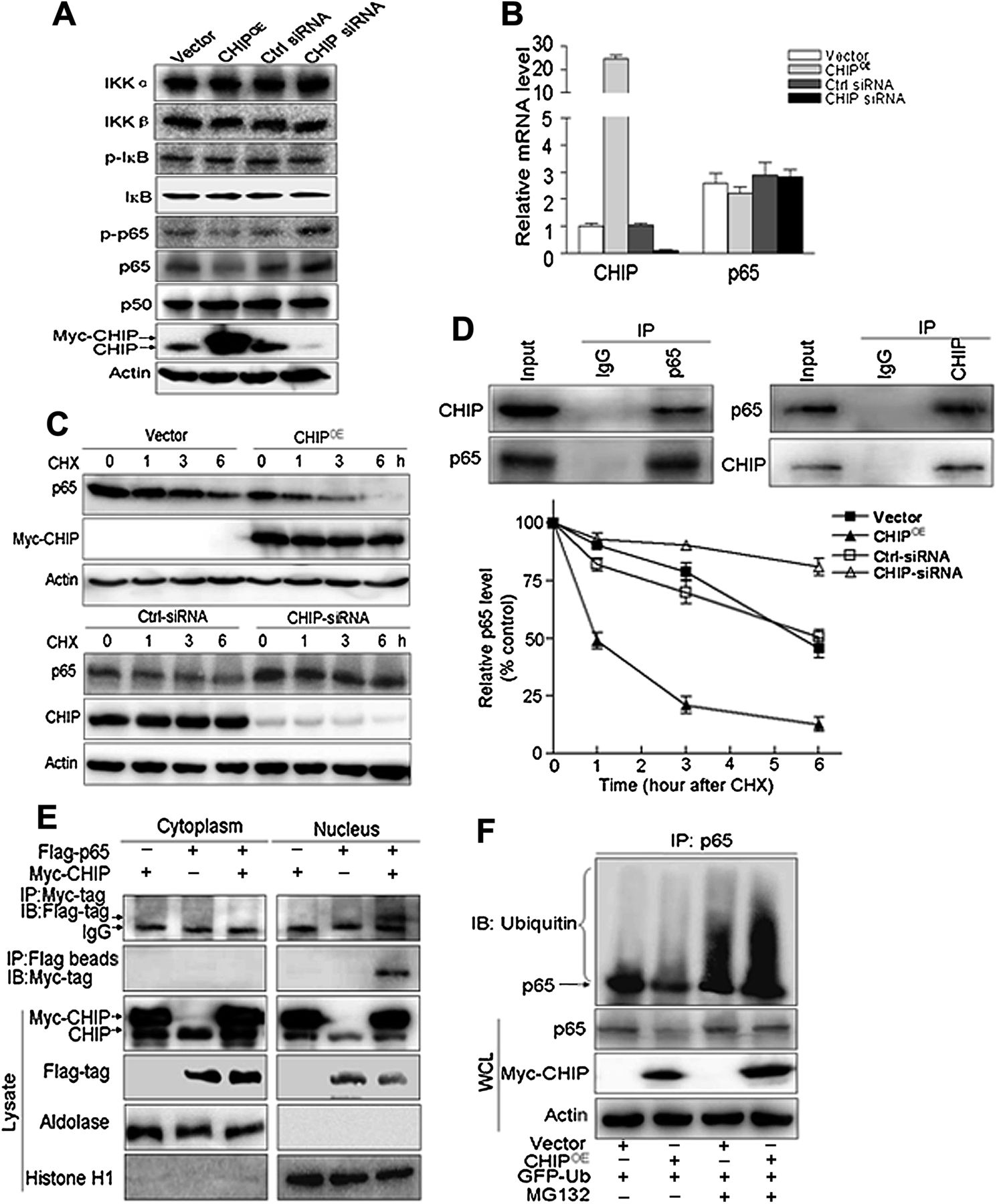
Furthermore, we investigated potential mechanisms of the NF-κB signalling pathway controlled by CHIP. There was no significant change in expression of IκB kinases (IKKα/β), phosphorylated IκBα (serine-32) or basic IκBα, and NF-κB/p50 between the CHIP expression modified and respective control cells (figure 5A). However, unexpectedly, NF-κB–p65 and its phosphorylated (serine-536) levels were decreased in CHIPOE cells, and increased in CHIP knockdown cells compared with corresponding control cells (figure 5A). In contrast, NF-κB–p65 mRNA level was not affected by CHIP overexpression or knockdown (figure 5B).

CHIP (carboxy terminus of Hsc70 interacting protein) promotes nuclear factor κB (NF-κB) ubiquitination for degradation by the proteasome. (A) The levels of protein in the canonical NF-κB activated signalling pathway were determined by immunoblotting in CHIP overexpressed (CHIPOE), CHIP small interfering RNA (siRNA) and their respective control (ctrl) transfected BGC823 cells. IκB, inhibitor of NF-κB; IKK, IκB kinases. (B) p65 mRNA expression is not affected by CHIP alteration in gastric cancer cells. Total mRNA was prepared from CHIPOE, CHIP siRNA and their respective control transfected BGC823 cells and p65 and CHIP mRNA levels were quantified using real time RT-PCR. (C) CHIP overexpression significantly enhanced but knockdown CHIP decreased the degradation of p65. BGC823 cells were transfected with CHIPOE or vector for 48 h and CHIP siRNA or ctrl siRNA for 72 h, respectively, followed by exposure to cycloheximide (CHX 50 mg/ml) for 0, 1, 3 or 6 h. Target proteins in whole cell lysates were detected by immunoblotting using antibodies against p65 and CHIP (left). The intensity of the p65 protein bands was analysed by densitometry, after normalisation to the corresponding β-actin level (right). The means ± SD are from three independent experiments. (D) BGC823 cells were pretreated with MG132 (10 μM) for 6 h and endogenous protein–protein interaction between CHIP and p65 was determined by immunoprecipitation (IP) with CHIP or p65 antibodies followed by immunoblotting. IgG was used as a negative control for immunoprecipitation. (E) Flag-p65 and Myc-CHIP were expressed in BGC823 cells with MG132 (10 μM) for 6 h. The cytoplasmic and nuclear extracts were immunoprecipitated using the anti-Myc antibody or anti-FLAG magnetic beads, and immunoblotted (IB) using the indicated antibodies. Aldolase and histone H1 were used as the cytoplasmic and nuclear loading controls, respectively. (F) Ubiquitination of p65 was induced by CHIP. GFP-ubiquitin (GFP-Ub) was coexpressed in BGC823 cells with myc-CHIP or vector control with or without treatment of MG132 (10 μM) for 6 h. Ubiquitinated p65 protein was immunoprecipitated using anti-p65 antibody and further detected with ubiqutin antibody. In the whole cell lysates, endogenous p65 and myc-CHIP were examined by anti-p65 and anti-myc antibodies.
We therefore postulated that CHIP may promote p65 destabilisation. When the cells were treated with cycloheximide, an inhibitor of protein synthesis, degradation of p65 was promoted by overexpression of CHIP whereas it was suppressed by CHIP knockdown (figure 5C). Additionally, a physical interaction between CHIP and p65 was determined by co-immunoprecipitation in both endogenous (figure 5D) and exogenous (myc-CHIP and Flag-p65) settings (figure 5E). Furthermore, co-immunoprecipitation experiments using cytoplasmic and nuclear fractions showed that Myc-CHIP interacted with Flag-p65 protein only in the nuclear extracts (figure 5E, right panel) but not from the cytoplasmic fraction (figure 5E, left panel).
It was also shown that loss of p65 expression in CHIPOE cells was inhibited by pretreatment with the proteasome inhibitor MG132 (figure 5F). Simultaneously, we found ubiquitinated p65 was increased due to MG132 inhibiting its degradation, and CHIP overexpression further enhanced the ubiquitination of p65 (figure 5F). These data indicated that CHIP functioned as an E3 ligase to mediate the ubiquitination of p65 for degradation by the proteasome.
U-box domain of CHIP is required for degradation of p65
To determine which motif of CHIP is required for degradation of p65, we transfected Myc-CHIP full length (CHIP-FL), TPR (CHIP-TPR) or U-box motif (CHIP-U-box) expression plasmid into BGC823 cells. We found that overexpression of the CHIP-U-box motif, as with CHIP-FL, reduced endogenous p65 levels but TPR motif overexpression had no effect (figure 6A). Consistent with this result, the ubiquitin conjugation of p65 was enhanced by overexpression of the U-box motif but not the TPR motif (figure 6B). We also elucidated which motif of CHIP binds to p65 by a co-immunoprecipitation assay. This revealed that the U-box motif, but not the TPR motif, retained the ability to associate with p65 (figure 6B).
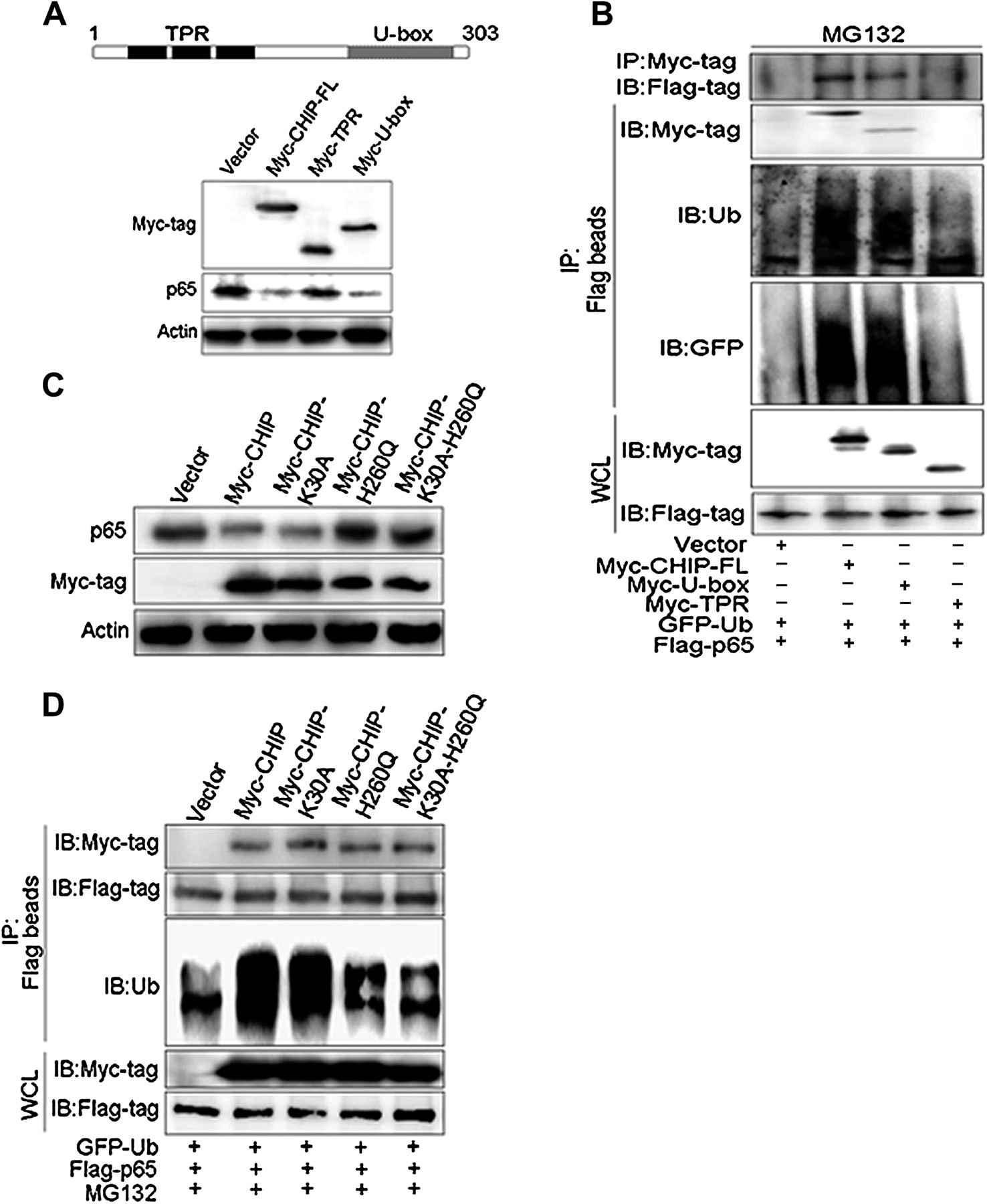
U-box domain of CHIP (carboxy terminus of Hsc70 interacting protein) is required for degradation of p65. (A) Two main functional domains of CHIP (tetratricopeptide repeat (TPR) and U-box) are schematically illustrated on top. BGC823 cells were transfected with Myc-CHIP-full length (FL) or its domains (Myc-TPR or Myc-U-box) for 48 h. The abundance of endogenous p65 and the efficiency for Myc-CHIPs transfection was examined by immunoblotting. (B) BGC823 cells were cotransfected with vector control, Myc-CHIP-FL, Myc-TPR and Myc-U-box, together with Flag-p65 and GFP-ubiquitin (GFP-Ub) for 48 h, then MG132 (10 μM) was added for the later 6 h. Co-immunoprecipitation (Co-IP) using anti-Myc antibody or anti-FLAG magnetic beads, and immunoblotting (IB) using the indicated antibodies were carried out to determine the interaction of CHIP and p65, and the ubiquitination of p65. (C) BGC823 cells were transfected with CHIP wild-type (Myc-CHIP), three mutants (Myc-CHIP-K30A, Myc-CHIP-H260Q, Myc-CHIP-K30A-H260Q) or vector control plasmid for 48 h. Expression of endogenous p65 protein was examined by IB. (D) BGC823 cells were transfected with CHIP wild-type, three mutants or vector control, together with Flag-p65 and GFP-Ub for 48 h, and MG132 (10 μM) was added for the later 6 h. Co-IP using anti-FLAG magnetic beads and IB using the indicated antibodies were carried out to determine the interaction of CHIP wild-type or mutants with p65, and the ubiquitination of p65.
Next we investigated whether the stability of p65 is associated with the activity of CHIP. For this we transfected CHIP K30A (abolish its chaperone role23), CHIP H260Q (abolish its E3 ligase activity35) and CHIP (K30A-H260Q) mutants into cells. We found that CHIP H260Q and CHIP (K30A-H260Q) mutants, but not CHIP K30A mutant, stopped loss of p65 levels (figure 6C) and caused a decrease in p65 ubiquitination, compared with wild-type CHIP (figure 6D). However, the co-immunoprecipitation assay showed that the amount of the three CHIP mutants pull down from exogenous p65 (Flag-p65) were not altered compared with the wild-type CHIP (figure 6D, upper panel), suggesting that neither K30 nor H260 of CHIP was required for the interaction of CHIP–p65. These data suggested that CHIP, through its U-box domain, mediated the degradation of p65, thus maintaining p65 protein at a low basal level, and this effect was unrelated to CHIP chaperone activity.
Expression of CHIP in gastric cancer cells inhibits angiogenesis in vivo
To further address the functional role of CHIP in angiogenesis, stable vector, CHIP-FL or truncated CHIP (TPR and U-box motif) BGC823 cells were generated and CHIP expression in these cells was confirmed by western blot (figure 7A). Tube formation assay showed that the average number of tubular structures formed by HUVECs was significantly decreased in conditioned medium from CHIP-FL and CHIP-U-box cells, but not from CHIP-TPR cells, when compared with vector control (figure 7B).

CHIP (carboxy terminus of Hsc70 interacting protein) overexpression in gastric cancer cells inhibits blood vessels formation in vivo. (A) Expression of CHIP-full length (FL) and two functional domains (Myc-U-box and Myc-tetratricopeptide repeat (TPR)) in BGC823 cells. Plasmid vector containing Myc-CHIP, Myc-U-box, Myc-TPR or empty vector control was transfected into BGC823 cells, which were selected with G418 (600 μg/ml). Exogenous CHIP (myc-CHIP) protein levels in the cells were analysed by immunoblotting with anti-myc antibody. (B) Representative images in the upper panel show CHIP-FL and CHIP-U-box but not CHIP-TPR expression in BGC823 cells inhibits human umbilical vein endothelial cell tube formation compared with vector control cells. In the right panel, numbers of tubes formed per field were counted in five random fields for each group. (C) Photograph of matrigel plugs excised from nude mice after 10 days of growth in vivo. (D) The extent of host angiogenesis was examined by immunofluorescent staining for expression of CD31 in matrigel plugs containing CHIP-FL and two truncated overexpressing or control BGC823 cells. DAPI nuclear staining indicated the overall cell density in each matrigel plug. (E) The number for CD31 positive cells was counted from five random fields for all vector control, CHIP-FL, CHIP-TPR and CHIP-U-box groups. (F) Expression of interleukin (IL)-8 mRNA was examined in matrigel plugs by real time RT-PCR (n=4/group). (G) Model of the CHIP suppresses tumour angiogenesis through the inhibition of nuclear factor κB (NF-κB) signalling. **p<0.01.
The in vivo matrigel plug assay was performed to further investigate whether CHIP expression could inhibit new blood vessel formation in nude mice. Visual examination revealed obviously less vascularisation in matrigel plugs containing CHIP-FL and CHIP-U-box cells than the vector control plugs whereas this effect was not observed in CHIP-TPR cells (figure 7C). CD31 staining also demonstrated that both of the CHIP-FL and CHIP-U-box plugs contained less neovessel density with a 50% reduction of CD31 positive cells (figure 7D,E). However, CHIP siRNA cells appeared more vascularised than control cells (see supplementary figure S4A,B, available online only), either by visual examination or with CD31 staining (see supplementary figure S4C,D, available online only). Quantitative RT-PCR demonstrated that IL-8 mRNA levels decreased significantly by 53% and 49% in matrigel plugs containing CHIP-FL and CHIP-U-box cells, respectively (figure 7F) whereas they were increased by 2.3-fold in CHIP siRNA plugs (see supplementary figure S4E, available online only) compared with the corresponding control plugs.
Tumour angiogenesis and metastasis is inhibited by CHIP expression in gastric cancer cells
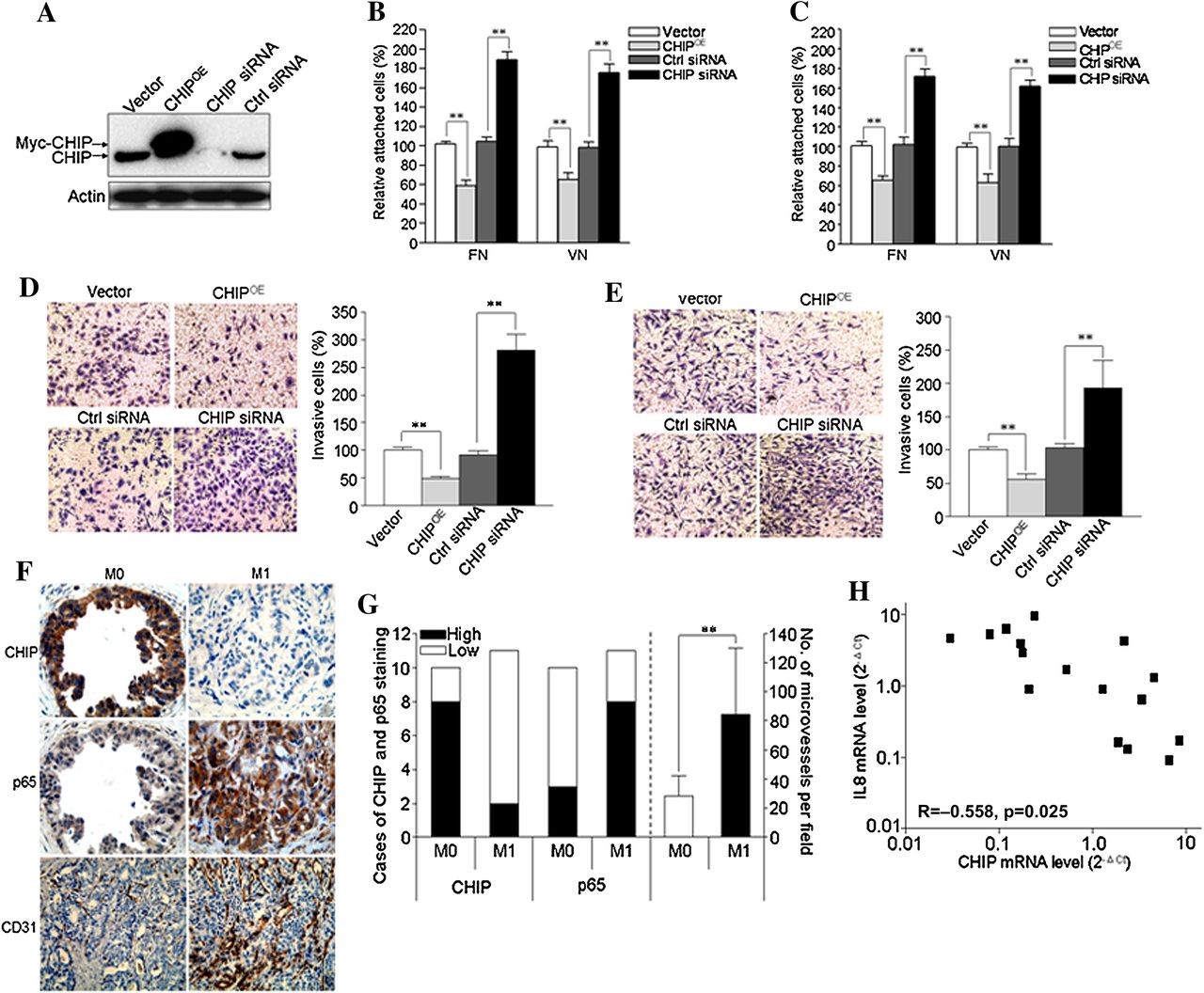
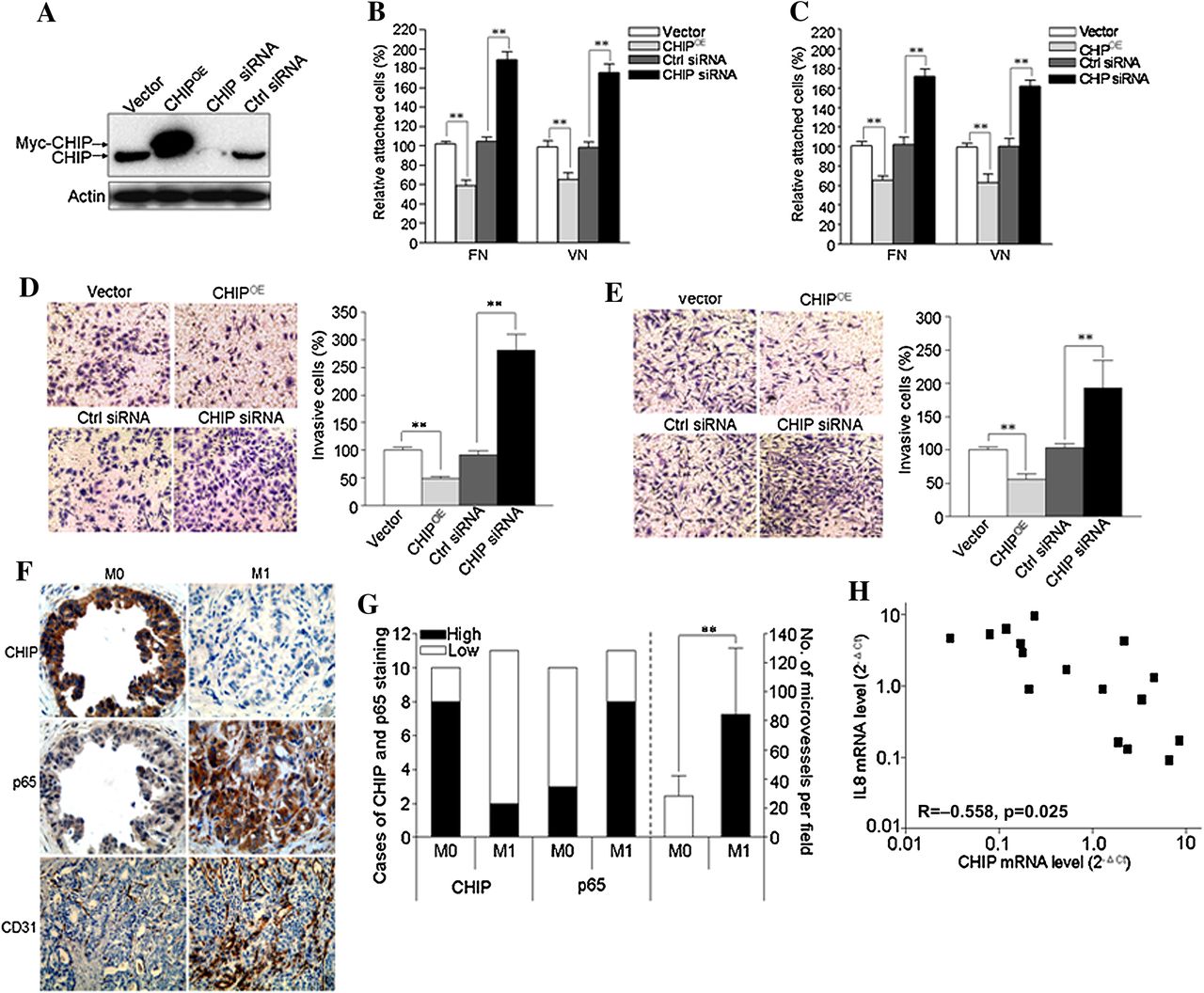
To examine the roles of CHIP on invasive and metastatic potential of GC cells, we performed adhesion and invasion assays. First, expression of CHIP was examined by western blot in CHIP overexpressed (CHIPOE) or knockdown (CHIP siRNA) BGC823 cells (figure 8A). In the adhesion assay, we found that CHIPOE decreased cell attachment ability of BGC823 cells on fibronectin and vitronectin coated plates by 41% and 32%, respectively, and CHIP knockdown increased this ability by 88% and 79%, respectively, compared with the corresponding controls (figure 8B). Moreover, similar results were confirmed in MGC803 cells (figure 8C).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CHIP (carboxy terminus of Hsc70 interacting protein) inhibits tumour angiogenesis and metastasis of gastric cancer (GC) cells. (A) BGC823 cells were transiently transfected with a plasmid expressing human CHIP protein (CHIPOE) or small interfering RNA (siRNA) to knockdown CHIP (CHIP siRNA) or their controls (vector or control (ctrl) siRNA, respectively). After 48 h, whole cell lysates were collected for detection of target proteins by immunoblotting. (B–C) 48 h after transfection, BGC823 cells (B) or MGC803 cells (C) were trypsinised and reseeded onto 96 well plates coated with fibronectin (FN) or vitronectin (VN), respectively. Cell attachment assay was carried out as described in the materials and methods section. (D–E) Cell invasion assay of BGC823 cells (D) and MGC803 cells (E) after CHIP overexpression or knockdown. Left panel shows the representative images of invaded GC cells in the insets of transwell chambers (original magnification, ×200). Right panel shows the quantification of cell invasion. Quantitation of the results was shown in the bar graph with means ± SD from three independent experiments. **p<0.01. (F–G) CHIP expression inversely correlates with p65 expression and microvessel counts by immunostaining for CD31. (F) Representative images of CHIP, p65 and CD31 staining in the primary tumour tissues of GC patients with distant or lymph node metastases (M1) and those without metastases (M0) (original magnification for CHIP and p65 in the same field, ×400; original magnification for CD31, ×200). (G) CHIP and p65 expression and the number of microvessels per field were quantified (M0, n=10; M1, n=11). **p<0.01. (H) Correlation of CHIP and interleukin (IL)-8 mRNA levels in the primary tumours of GC patients (n=16).
We also investigated the invasiveness of these cells, and found BGC823 and MGC803 CHIPOE cells had significantly decreased ability to penetrate the matrigel coated membrane by 51% and 48%, respectively, whereas silencing of CHIP in BGC823 and MGC803 cells increased the invasive ability by 175% and 90%, respectively, compared with the corresponding controls (figure 8D,E).
To examine the role of CHIP on tumour angiogenesis and subsequent metastasis, six GC cell lines were used. It was shown that CHIP expression inversely correlated with IL-8 expression in GC cells (see supplementary figure S5A,B, available online only). The cells with lower CHIP expression promoted HUVEC tube formation and exhibited higher cell invasion potential (see supplementary figure S5C,F, available online only). To confirm these results, CHIP and p65 expression and microvessel count were determined in the primary tumours of GC patients with or without metastases (M1 or M0). We found M0 tumours revealed higher CHIP expression but lower p65 expression and microvessel density compared with those in the M1 group (figure 8F,G). Moreover, CHIP mRNA level inversely correlated with IL-8 mRNA level in the primary tumours of GC patients (correlation coefficient (R) =−0.558, p=0.025; figure 8H).
Discussion
Metastatic GC is a life threatening disease with no effective clinical treatment. Therefore, it is important to understand the molecular mechanisms of GC progression in order to discover new potential treatment targets.
CHIP is a ubiquitin ligase with tumour suppressor effects by inducing ubiquitination and degradation of numerous oncogenic proteins.21–25 ,28 ,29 Despite this, there are few studies exploring CHIP expression related to progression and prognosis of cancer. Jan et al reported that decreased CHIP expression is correlated with unfavourable tumour grade, later pathological stage, larger tumour size and poor disease free survival in breast cancer patients.30 In the present study, we showed that CHIP expression was lower in gastric cancerous tissues compared with non-cancerous tissues. Moreover, low expression of CHIP was significantly associated with unfavourable clinicopathological variables and shorter survival in gastric carcinoma patients, which was identified in two independent cohorts. In addition, multivariate analysis indicated that CHIP was an independent prognostic indicator of gastric carcinoma. Furthermore, CHIP risk score significantly increased the prognostic value of TNM stage, histological type and tumour diameter. These findings indicate that CHIP may be involved in the progression of gastric carcinomas and be a significant prognostic indicator for gastric carcinoma patients.
Our clinical data urged us to perform a series of cell line and animal experiments to explore the possible mechanisms responsible. Several studies have shown that tumour associated neovasculature is closely related to tumour growth and metastasis in various tumours, including breast, lung, prostate, colorectal as well as GC.36–39 Our xenograft studies in immunocompromised nude mice indicated that the tumours from the CHIP overexpression group had reduced blood vessel formation, suggesting CHIP may suppress angiogenesis in GC. Angiogenesis is a multistep process, which includes endothelial cell proliferation, migration and the formation of blood vessels.40 In this study, we found that both the growth and tube formation of HUVECs were inhibited in conditioned medium collected from CHIP overexpressing GC cells. Furthermore, we demonstrated that CHIP overexpression in GC cells inhibited the supportive vasculature in vivo. In addition, we showed that overexpression of CHIP suppressed GC cell adhesion and invasion, which are two key events of tumour metastasis, consistent with its role in other malignant cells such as breast cancer29 and lung cancer.28 Moreover, we found that tumours from patients without metastases revealed higher CHIP expression but lower p65 expression and microvessel density compared with those in the metastases group. Based on these findings and combined with the fact that metastasis is the major cause of GC patient death, it is not surprising to see that reduced CHIP is associated with tumour angiogenesis induced tumour invasion and metastasis, and eventually worse outcome for GC patients.
Activated NF-κB has a crucial role in cancer development and progression and it regulates downstream genes, including IL-6, IL-8, MMP-2, VEGF and cyclooxygenase 2 to promote proliferation, survival, angiogenesis and metastasis.10 ,41 Our study showed that CHIP regulated the protein level of the p65 subunit of NF-κB but not the p50 subunit, and subsequently inhibited NF-κB DNA binding activity. Moreover, CHIP overexpression suppressed expression of NF-κB downstream genes, especially IL-8. Clinical studies have shown that IL-8 is upregulated in several human malignancies, including melanoma,42 colon,43 non-small cell lung44 as well as GC.45 Moreover, IL-8 production is linked to tumour vascularisation, metastatic phenotype and an overall poor prognosis.39 ,42 ,46 We found CHIP overexpression decreased, whereas CHIP knockdown increased, both IL-8 mRNA expression and protein secretion. The role of IL-8 in CHIP regulated GC angiogenesis was further confirmed by IL-8 rescue and blocking assays. Therefore, the reduction of IL-8 protein secreted by GC cells seemed to account for the inhibitory effect of CHIP on GC angiogenesis.
IκBα mediated cytoplasmic accumulation of the NF-κB complex has been considered a key mechanism of terminating NF-κB signalling.47 ,48 However, CHIP did not affect the activation and expression of IκBα in our study. In addition, several recent reports have indicated that ubiquitin and proteasome dependent degradation of the NF-κB family member p65/RelA in the nucleus is also important for efficient and prompt termination of NF-κB activation.14–18 In this study, we unexpectedly found that CHIP could interact with p65 in the nucleus but not in the cytoplasm, and acted as ubiquitin E3 ligase that targeted p65/RelA degradation and terminated NF-κB activation. This was consistent with previous reports that identified CHIP interacting with nuclear Runx127 and Runx249 and mediating their degradation via the ubiquitin–proteasome pathway.
Taken together, the data presented here showed that loss of CHIP expression was significantly correlated with GC progression and was an independent prognostic marker of poor overall survival in GC patients as well as adding significant prognostic value to the well known clinical prognostic factors. CHIP inhibited GC cell malignant phenotypes and, importantly, suppressed GC angiogenesis by inhibiting NF-κB activity through ubiquitin–proteasome dependent degradation of the NF-κB–p65 and downregulation of the proangiogenic cytokine IL-8. As NF-κB–p65 target hundreds of downstream genes, it is possible that several other genes may be involved in this process. Based on this study, we propose that CHIP may serve as a promising prognostic marker for GC, and restoration of CHIP may be a novel strategy for antiangiogenesis therapy for human GC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
-
Funding This study was supported in part by the project funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, the National Natural Science Foundation of China (30930080, 81161120537 to JWZ, 81001231 to SW) and the Postdoctoral Science Foundation of China (20100481165 to SW).
-
Competing interests None.
-
Ethics approval The use of human gastric cancer tissues and the waiver of patient consent in this study were approved by the Clinical Research Ethics Board of the Nantong Cancer Hospital and the Ethics Committee of the Nanjing Medical University, respectively. The study was conducted according to the principles expressed in the Declaration of Helsinki.
-
Provenance and peer review Not commissioned; externally peer reviewed.