Article Text

Abstract
Objective The anti-leukemic drugs, azathioprine and 6-mercaptopurine (6MP), are important in the treatment of inflammatory bowel disease but an alternative faster-acting, less-allergenic thiopurine, 6-thioguanine (6TG), can cause hepatic veno-occlusive disease/sinusoidal obstructive syndrome (SOS). Understanding of SOS has been hindered by inability to ethically perform serial liver biopsies on patients and the lack of an animal model.
Design Normal and C57Bl/6 mice with specific genes altered to elucidate mechanisms responsible for 6TG-SOS, were gavaged daily for upto 28d with 6TG, 6MP or methylated metabolites. Animal survival was monitored and at sacrifice a histological score of SOS, haematology and liver biochemistry were measured.
Results Only 6TG caused SOS, which was dose related. 6TG and to a lesser extent 6MP but not methylated metabolites were associated with dose-dependent haematopoietic toxicity. SOS was not detected with non-lethal doses of 6TG. SOS did not occur in hypoxanthine-phosphoribosyl transferase-deficient C57Bl/6 mice, demonstrating that 6TG-SOS requires thioguanine nucleotides. Hepatic inflammation was characteristic of SOS, and C57Bl/6 mice deficient in P- and E-selectins on the surface of vascular endothelial cells showed markedly reduced SOS, demonstrating a major role for leukocytes recruited from blood. Split dosing of 6TG markedly attenuated SOS but still effected immunosuppression and prevented spontaneous colitis in Winnie mice, which have a single nucleotide polymorphism mutation in Muc2.
Conclusion This novel model provides clinically relevant insights into how 6TG induces SOS, and how this dangerous adverse drug reaction may be avoided by either inhibition of endothelial activation or simple changes to dosing regimens of 6TG, while still being effective treatment for colitis.
- Thioguanine
- IBD
- veno-occlusive disease
- sinusoidal obstructive syndrome
- nodular regenerative hyperplasia
- 6-mercaptopurine
- hepatic iron metabolism
- Crohn's disease
- mucosal infection
- mucus
- mucins
- gastric mucosal barrier
- enteric infections
- colonic bacteria
- azathioprine
- mucosal barrier
- dendritic cells
Statistics from Altmetric.com
- Thioguanine
- IBD
- veno-occlusive disease
- sinusoidal obstructive syndrome
- nodular regenerative hyperplasia
- 6-mercaptopurine
- hepatic iron metabolism
- Crohn's disease
- mucosal infection
- mucus
- mucins
- gastric mucosal barrier
- enteric infections
- colonic bacteria
- azathioprine
- mucosal barrier
- dendritic cells
Significance of this study
What is already known on this subject?
-
Thiopurine drugs are associated with the serious adverse drug reactions known as hepatic veno-occlusive disease/sinusoidal obstructive syndrome (SOS) and nodular regenerative hyperplasia.
-
There is controversy in the literature about whether this is a generalised effect of all thiopurine drugs or an idiosyncratic 6-thioguanine (6TG) effect.
What are the new findings?
-
SOS is not a thiopurine class effect in this novel mouse model.
-
SOS is dependent on 6TG dose.
-
Thioguanine nucleotides are required for the pathogenesis of 6TG-SOS.
-
Inflammation plays a critical role in the pathogenesis of 6TG-SOS.
How might it impact on clinical practice in the foreseeable future?
-
Drugs that inhibit hepatic sinusoidal endothelial activation, and thereby impair recruitment of circulating leucocytes, may prevent SOS.
-
A dosing regimen of 6TG which reduces the concentration of thioguanine nucleotides in the hepatic circulation would prevent SOS but still be effective treatment for inflammatory bowel disease.
Introduction
Azathioprine (AZA) and the closely related drug 6-mercaptopurine (6MP) are anti-neoplastic prodrugs which use the salvage pathway of purine metabolism (figure 1) to form thioguanosine triphosphate (TGTP). TGTP competes with guanosine triphosphate (GTP) via GTP-binding proteins in activated T lymphocytes to effect immunosuppression.2 These prodrugs have become a cornerstone of maintenance therapy for clinically moderate to severe Crohn's disease and ulcerative colitis because they are effective and economical.3–5 Furthermore, AZA provides additional benefit when prescribed as co-therapy with anti-tumour necrosis factor α therapy for Crohn's disease.6

Thiopurine prodrugs are metabolised via the purine salvage pathway. Thiopurine (pro-) drugs: azathioprine (AZA), 6-mercaptopurine (6MP), 6-thioguanine (6TG). Anabolic enzymes and metabolites: inosinemonophosphate dehydrogenase (IMPDH), guanosinemonophosphate synthetase (GMPS), hypoxanthine guanine phosphoribosyl transferase (HPRT), thioinosine monophosphate (TIMP). Thioguanine nucleotides: thioguanosine monophosphate (TGMP), thioguanosine diphosphate (TGDP), thioguanosine triphosphate (TGTP). Methylation enzymes and metabolites: thiopurine S-methyltransferase (TPMT), methyl-6-mercaptopurine (me6MP), methyl-6-mercaptopurine ribonucleoside (me6MPr), methyl-6-thioguanine (me6TG), methyl-6-thioinosine monophosphate (meTIMP). Oxidative enzymes and metabolites: xanthine oxidase (XO), aldehyde oxidase (AO), guanase, thiouric acid(TUA). AZA is non-enzymatically converted to 6MP in the small intestine by a glutathione-dependent reaction. 6MP and 6TG are metabolised via catabolic pathways to TUA and methylation products, and via purine salvage to TGMP and thence via phosphorylation kinases to TGDP and TGTP. TGTP is the principal active drug. IMPDH is rate limiting and subject to product inhibition.1
However, the immunomodulatory doses of AZA or 6MP used to treat inflammatory bowel disease (IBD) have a very slow onset of therapeutic action (3–4 months) and are eventually ineffective in about 40% of patients with IBD as a result of poor efficacy and/or adverse drug reactions (ADRs).7 Some ADRs are associated with methylated by-products of 6MP metabolism,8 ,9 and we and others have shown that some dose-related ADRs may be avoided by prescribing low-dose 6MP or AZA in combination with allopurinol, a manoeuvre which is associated with less methylated by-products.1 ,10 ,11 However, idiosyncratic ADRs are not prevented by this manoeuvre.
6-thioguanine (6TG), an analogue of guanine, is a prodrug also metabolised to TGTP (figure 1). 6TG has a pharmacodynamic action measured within weeks rather than the typical 4 months for AZA or 6MP,12 ,13 and does not lead to production of methylated 6MP metabolites,14 ,15 or the idiosyncratic ADRs associated with AZA and 6MP. These properties of 6TG—faster action and fewer ADRs—should make it the preferred thiopurine drug. However, 6TG can cause hepatic veno-occlusive disease (VOD)16 and nodular regenerative hyperplasia (NRH).17 As a result of concern about the risk of VOD or NRH, prescribing of 6TG for IBD has been tentative, even though it was found effective in open-label studies of patients with IBD not responding to AZA or 6MP.12–15
VOD and NRH are part of a spectrum of vascular disorders of the liver caused by thrombotic and/or endothelial injury of the microvasculature.18–20 The first reported example of VOD was Jamaican tea disease, caused by monocrotaline, a compound in Senecia tea. There is a well described rat animal model for monocrotaline-induced VOD in which it has been shown that the earliest insult is the vascular pathology known as hepatic sinusoidal obstructive syndrome (SOS). Sinusoidal endothelial cells, the initial target in SOS, embolise and obstruct the hepatic microcirculation to cause VOD.21–23
In this study we report on a novel murine model of SOS due to 6TG. We show in the model that the SOS pathology is 6TG dose related, does not occur with a cumulative small dose, is not a thiopurine class effect, is mediated by thioguanine nucleotides (TGNs) and not the prodrug, is amplified by inflammatory leucocytes entering the liver, and can be avoided by a manoeuvre that reduces the concentration of drug in the portal circulation.
Material and methods
Animals, ethics and drugs
Groups of n=6, 6–10-week-old male or female wild type (WT), hypoxanthine (guanine) phosphoribosyl transferase (Hprt) deficient, PE-selectin −/− and Winnie C57Bl/6 mice were intra-gastrically gavaged daily with 100 μl of tap water (vehicle control) or drugs in the same volume of water for periods between 3 and 28 days. The drugs were 6TG, 6MP, methylated 6MP (me6MP) or methylated 6TG (me6TG) (≤5 mg/kg/d) (Sigma, Castle Hill, Australia). Mice were monitored and scored daily. Mice were sacrificed if body weight fell more than 20% compared with day 0 or if they scored more than 4 for body weight, habitus and behaviour (activity, hunching). All experiments were performed in accordance with the protocols approved by the University of Queensland Animal Ethics Committee.
After blood collection for biochemistry and peripheral blood count, animals were sacrificed by cervical dislocation. Liver was fixed in 20% formalin for haematoxylin–eosin or periodic acid Schiff (PAS) staining, F4/80 (Abcam, Cambridge, UK) and von Willebrand factor (vWF) (Abcam) and terminal deoxynucleotidyl transferase mediated dUTP nick end labelling assay (TUNEL) immunofluorescence (Roche staining kit, Welwyn Garden City, UK). Liver for ultrastructural study was fixed in 4% glutaraldehyde and stored in 0.1 M cacodylate buffer. Liver sections for mRNA were frozen and stored at −80°C. One femur was also collected for bone marrow cellular count: the bone was flushed at proximal and distal ends with phosphate-buffered saline +2% fetal calf serum. Cells were counted and viability assessed using 0.4% Trypan blue.
SOS scoring
Development of the scoring system was initially informed by the rat monocrotaline induced SOS model and included the main characteristics previously described in that model21: damage of the endothelial lining of the central vein; subendothelial and sinusoidal haemorrhage; coagulative necrosis; subendothelial fibrosis; inflammation due to monocytes/macrophages and neutrophils. Scoring of these criteria was applied to pilot experiments which were performed in 6-week C57Bl/6 male and female mice treated with 6TG 2.5–5 mg/kg/day up to 12 days (data not shown). Histological assessment revealed some traits which were common and others different from the monocrotaline-induced SOS. Similar traits were: damage of endothelial lining of central veins, sinusoidal congestion with erythrocytes, necrosis and inflammation. Histological traits not reported in the monocrotaline model were: sinusoidal dilatation, central vein sludging with cellular debris, microvesicular steatosis, and apoptosis. Traits not seen in the 6TG-treated mice but reported in the monocrotaline model were fibrosis and subendothelial haemorrhage. The final scoring system developed by two of the authors (IO and RL) included all the traits recorded in the pilot experiments, comprising three subscores related to sinusoidal damage, central vein damage and necroinflammatory damage (table 1). Scoring was performed in a blinded fashion for 10 central veins in three to four low-power (20×) fields. The maximum possible SOS score was 240, being the sum of the sinusoidal damage subscore (maximum 60), central vein damage subscore (60) and parenchymal necroinflammatory subscore (120). All scoring was performed by the first author (IO). Intra-observer variability was assessed in six vehicle control and six test livers over three scoring runs, each more than 1 month apart. Mean coefficient of variance was 8.8%, range 4–13%.
Sinusoidal obstructive syndrome subscores
Colitis scoring, immunochemistry, RNA extraction, cDNA synthesis and measurement of TGNs
These were performed as previously described.24 ,25 VE-cadherin and β-actin (online supplementary figure 1) were used for housekeeping mRNA genes. Oligomer nucleotide sequences are shown in supplementary figure 2. TGNs were measured in supernatant using the Dervieux-Boulieu method26 and corrected to the liver or packed blood cell pellet weight.
Hprt-deficient mice breeding and genotyping
Breeding-age C57Bl/6 female mice homozygous for Hprt were paired with hemizygous-deficient mutant male mice to generate Hprt-deficient mice. Offspring were genotyped to confirm Hprt deficiency (online supplementary figure 3) using the Hprt primers shown in online supplementary figure 1.
Transmission electronic microscopy
Transmission electronic microscopy samples were analysed using a JEOL transmission electronic microscopy (Tokyo, Japan). Endothelial cell tight junctions were quantified by counting the tight junctions from 10 high-power fields in vehicle control and 6TG 2.5 mg/kg/day C57Bl/6 mice.
Statistical analysis
Comparison of groups was performed using appropriate non-parametric tests (Mann–Whitney U test, analysis of variance, Kruskal–Wallis test) and the log-rank Mantel–Cox test for survival analyses, and were considered significant for two-tail at p<0.05. The results are presented as box and whisker plots showing median, quartiles and range, or as column graphs of mean ± SD.
Results
Daily gavage of high-dose 6TG, but not 6MP, impairs survival and damages the liver and bone marrow in C57Bl/6 mice
VOD-like illness has not developed in previous studies of rodents treated with 6TG.27 These studies administered 6TG in drinking water, whereas in our experiments we controlled the amount of 6TG administered by using bolus oral gavage dosing. When given as a daily oral gavage, 6TG was lethal at ≥1 mg/kg/day (figure 2A). However, mice gavaged with the lower dose of 0.5 mg/kg/day 6TG or with 2.5 mg/kg/day 6MP had 100% survival as did controls gavaged with vehicle only. Body weight (figure 2B), liver weight (figure 2C), cellularity in bone marrow (figure 3A) and peripheral blood leukocytes–lymphocytes and haemoglobin were reduced with increasing 6TG daily gavage dose over 28 days (figure 3B–D). 6MP was not associated with reduced total body or liver weight (figure 2B,C). Bone marrow cell, haemoglobin and total leukocyte–lymphocyte counts in venous blood were decreased with 6MP consistent with a mild immunosuppression (figure 3).
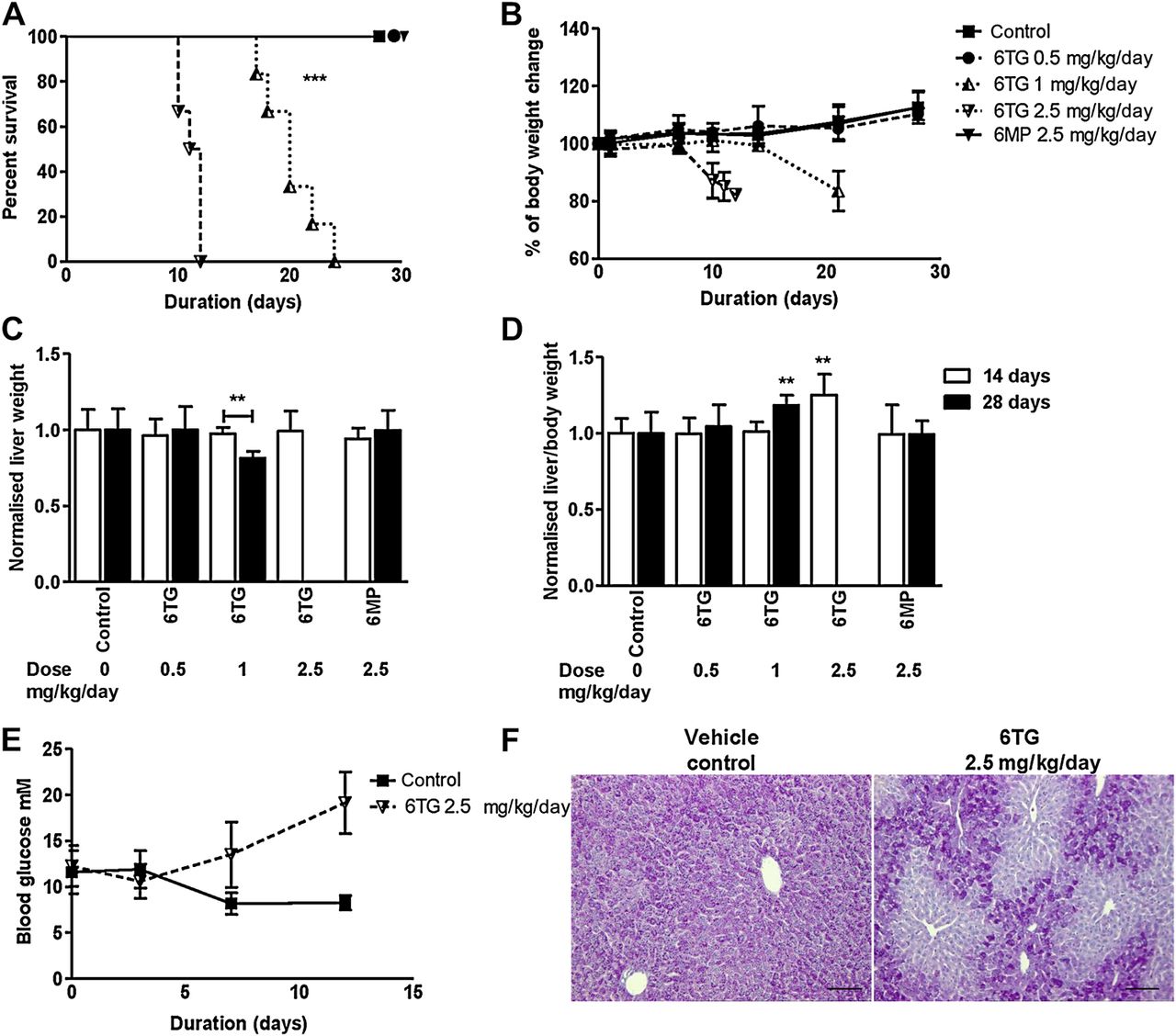
Daily gavage administration of high-dose 6-thioguanine (6TG) impaired survival and reduced body and liver weight. C57Bl/6 mice (female mice, n=6 in each group) were gavaged daily for up to 28 days with 6TG (0.5, 1 and 2.5 mg/kg/day) or 6-mercaptopurine (6MP) (2.5 mg/kg/day). (A) Survival decreased with the higher doses of 6TG (1 and 2.5 mg/kg/day, log-rank Mantel–Cox test ***p<0.001). (B) Body weight decreased with the higher 6TG doses of 1.0 and 2.5 mg/kg/day (Mann–Whitney vs control **p<0.01). (C) Liver weight normalised to vehicle-only controls in 14-day and 28-day experiments, was decreased only in the 6TG mice gavaged 1 mg/kg/day in the 28-day experiment. (Mice gavaged 2.5 mg/kg/day 6TG did not survive to 14 days.) (D) Liver weight relative to animal body weight at sacrifice was increased with 2.5 mg/kg/day 6TG in the 14-day experiment, and with 1 mg/kg/day 6TG in the 28-day experiment. (Mann–Whitney vs respective controls ** p<0.01). (E) Mean (SD) blood glucose in n=4 control and n=4 2.5 mg/kg/day 6TG male C57Bl/6 mice (two-way analysis of variance p=0.0017). (F) Representative histology, periodic acid Schiff (PAS) stain, showing glycogen depletion with 6TG 2.5 mg/kg/day for 10 days.
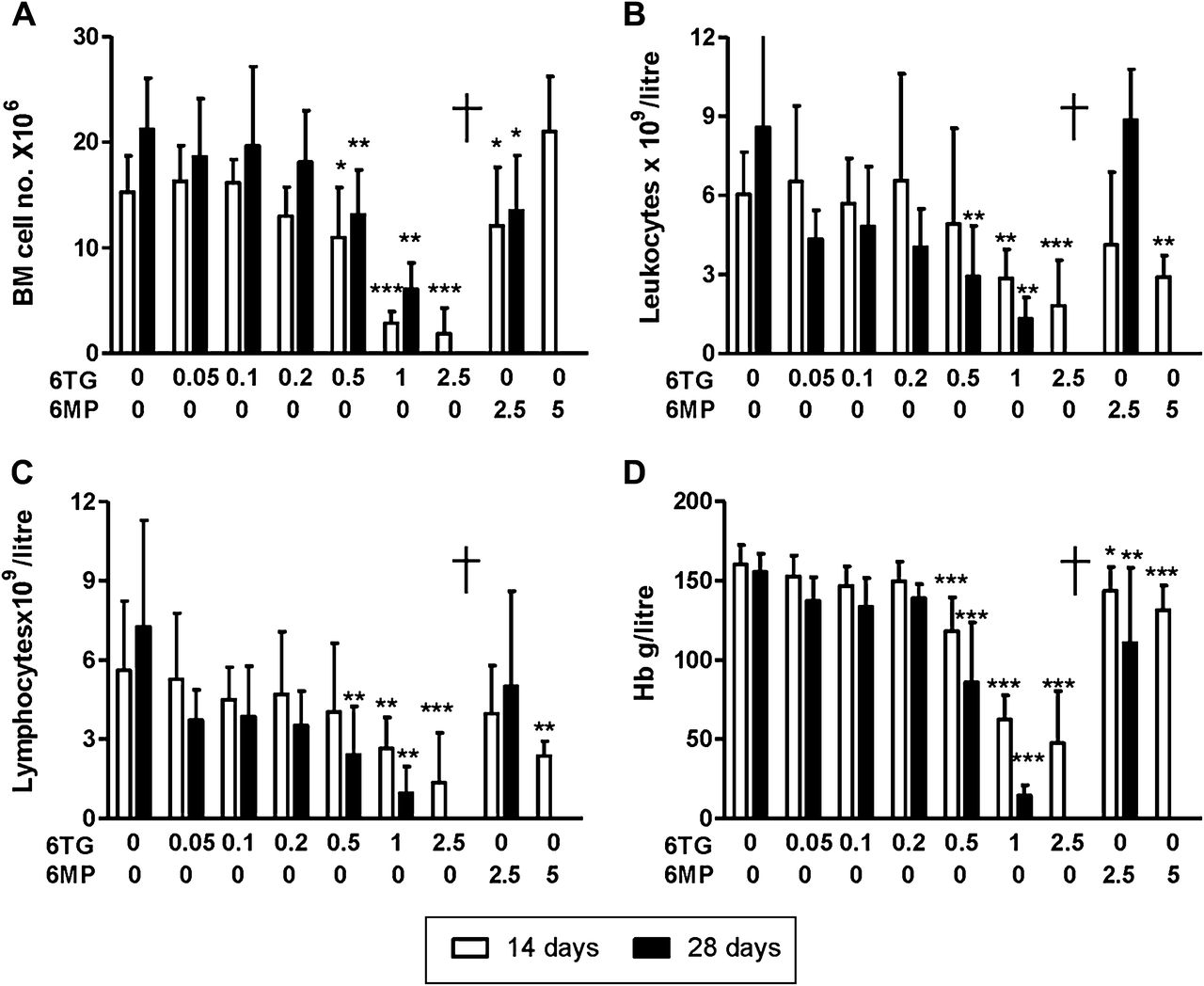
6-thioguanine (6TG) and to a lesser extent 6-mercaptopurine (6MP) were associated with dose-dependent haematopoietic toxicity. Once daily gavage administration of vehicle, 6TG (0.05, 0.1, 0.2, 0.5, 1 or 2.5 mg/kg/day) or 6MP (2.5 or 5 mg/kg/day) for 14-day and 28-day experiments in C57Bl/6 mice. Columns represent means and SDs for 14 and 28 days. (A) Bone marrow haematopoietic cells from one femur were decreased with 6TG ≥0.5 mg/kg/day and to a lesser extent with 6MP (6MP 5 mg/kg/day was tested only over 14 days). Total leucocyte counts (B), lymphocyte counts (C) and haemoglobin concentration (D) in venous blood from the same experiments. †= no data due to reduced survival. *p<0.05, **p<0.01, ***p<0.001.
In addition to the morbidity associated with immunosuppression, animal body weight decreased with 6TG gavage ≥1 mg/kg/day (figure 2B), which suggested that survival in these experiments was influenced by other factors such as impaired nutrition or metabolism. We measured glucose in non-fasting control and 6TG 2.5 mg/kg/day gavaged mice and found mean venous blood glucose (mmol/litre) increased in 6TG gavaged mice (figure 2E). We therefore assessed hepatic glycogen stores, which were depleted in these mice when killed (figure 2F). Depletion of glycogen stores was also noted with 6TG doses of 1 mg/kg/day for 14 days, but the depletion was restricted to the peri-central vein location. Glycogen depletion was not detected with 0.5 mg/kg/day. Despite reduction of glycogen stores—for every mole of glycogen there are three moles of water28—there was a 6TG dose-dependent increase in liver weight normalised to total body weight (figure 2D). The relative increase in liver weight was probably due to a combination of congestion and microsteatosis and the influx of inflammatory cells (figure 4A).

6-thioguanine (6TG) but not 6-mercaptopurine (6MP) causes sinusoidal obstructive syndrome (SOS). Daily gavage of 6TG 2.5 mg/kg/day for up to 12 days, but not vehicle control or 6MP 2.5 or 5 mg/kg/day for 14 days, resulted in SOS in C57Bl/6 wild type (WT) mice. (A) Representative histology from liver sections of a C57Bl/6 mouse gavaged with 6TG versus a C57Bl/6 mouse gavaged with vehicle or 6MP 5 mg/kg/day illustrates characteristics consistent with SOS. Bar scale 100 μm. Toluidine blue stain: showing sinusoidal dilatation around a ragged central vein with 6TG; von Willebrand factor immunohistochemistry (vWF): showing loss of endothelium with 6TG; haematoxylin and eosin (H&E) histology: showing congestion and central vein sludging with 6TG; F4/80 immunohistochemistry: revealing a marked macrophage infiltrate with 6TG; terminal deoxynucleotidyl transferase mediated dUTP nick end labelling assay (TUNEL) immunofluorescence: showing apoptosis. (B) Sinusoidal damage, central vein and necroinflammatory subscores, and total SOS score demonstrate SOS with 6TG but not with 6MP. Statistics: n=6 C57Bl/6 WT mice from each of vehicle only, 6MP 2.5 or 5 mg/kg/day gavaged once daily for 14 days, 6TG 2.5 mg/kg/day for 9–12 days. Box and whisker plots of median, quartiles and range. Mann–Whitney **p<0.01 for 6TG versus all other groups.
6TG but not 6MP causes SOS
We undertook histological examination of the livers of animals treated with 2.5 mg/kg/day 6TG. This revealed pathology characterised by damage to sinusoids, central veins and parenchyma in the treated animals. Figure 4A illustrates clear histological differences between C57Bl/6 mice gavaged vehicle or 6MP 5 mg/kg/day for 14 days and 6TG 2.5 mg/kg/day for 12 days. Liver histology of 6TG gavaged mice typically showed dilated sinusoids easily identified in toluidine blue stained thin sections; loss of endothelium around central veins demonstrated by absence of vWF staining of endothelial cells; sinusoidal congestion, central vein sludging, and microsteatosis observed after haematoxylin and eosin (H&E) staining; neutrophil and macrophage infiltrates in the parenchyma seen with both H&E staining and F4/80 staining for macrophages; and increased apoptosis as demonstrated with TUNEL staining (figure 4A). The increased apoptosis occurred in hepatocytes and endothelial cells as demonstrated by TUNEL, 4',6-diamidino-2-phenylindole (DAPI) and vWF co-staining (online supplementary figure 4). These findings are consistent with SOS.22 ,23
All SOS subscores (table 1) were increased with 6TG 2.5 mg/kg/day (figure 4B). The sinusoidal damage subscore appeared most increased with a median 63% (of the maximum possible 100%), central vein damage subscore was 52%, and the necroinflammatory subscore was 38% (figure 4B). However, daily gavage of 6MP at doses which caused mild haematopoietic suppression (figure 3) did not result in detectable SOS (figure 4B).
6TG-induced SOS is dose related
The current literature is controversial about whether 6TG-associated SOS is idiosyncratic or dose related.17 ,29–31 However, our mouse model data showed a dose-related response. SOS occurred incrementally in the liver with 6TG doses of 0.5–2.5 mg/kg/day for up to 14 days (figure 5A). Gavage administration of lower doses of 6TG (0.05–0.5 mg/kg/day) for 28 days caused minor sinusoidal dilatation and inflammation but did not cause SOS (figure 5B). The total SOS score for 28 days (13.0±8.6) was not greater than the SOS score at 14 days (15.6±7.7) with 6TG 0.5 mg/kg/day.

6-thioguanine (6TG)-induced sinusoidal obstructive syndrome (SOS) is dose dependent. (A) SOS occurred in a dose-dependent fashion with 6TG ≥1 mg/kg/day once daily gavages in C57Bl/6 wild type (WT) mice. n=6 in each group: 6TG 0, 0.5 or 1 mg/kg/day for 14 days and 2.5 mg/kg/day for median 11 days. Statistics: Mann–Whitney: **p<0.01, ***p<0.001. (B) SOS did not occur with once-daily gavaging of non-lethal doses of 6TG ≤0.5 mg/kg/day for 28 days. Graphs: columns show means ± SD, Mann–Whitney 0.1 mg/kg/day versus control, p=0.06, other doses versus control, p>0.2. (Note: central vein subscores were all zero.)
SOS and immunosuppression are due to thioguanine nucleotides
Stork et al,16 who reported VOD in children treated with anti-leukemic doses of 6TG, hypothesised that VOD could arise from 6TG itself or another 6TG metabolite such as me6TG. With respect to the ADRs of (non-VOD) hepatitis, it was hypothesised that me6MP or me6MP ribosides, which are associated with the hepatitis, are themselves causative.8 We investigated these possibilities by daily gavaging supra-pharmacological doses of me6MP or me6TG for 14 days. 6TG caused SOS, but 6MP, me6MP or me6TG did not (figure 6A). 6TG decreased body weight (p=0.004) as expected, but 6MP, me6MP and me6TG did not affect body weight (data not shown). 6TG decreased peripheral blood leucocytes, 6MP had less effect at 14 days, but me6TG and me6MP had no significant effect on leucocyte counts (figure 6B). Liver transaminases measured in venous blood at sacrifice were elevated with daily 6TG gavage of 2.5 mg/kg/day: median aspartate transaminase was increased 11-fold (p=0.02) and median alanine transaminase fourfold (p=0.03). Median alkaline phosphatase was not higher (p=0.2). Gavage with 6MP, me6MP or me6TG was not associated with increased liver enzymes, and liver enzyme levels were not altered in other experiments with doses of 6TG ≤1 mg/kg/day for shorter or longer periods of gavage (data not shown). This pattern of liver biochemistry is consistent with clinical findings with acute SOS-VOD or NRH in humans, in whom blood liver biochemistry is found to be significantly abnormal only with high-dose cytotoxic drugs23 but not lower immunomodulatory doses of 6TG.

Sinusoidal obstructive syndrome (SOS) and immunosuppression are caused by thioguanine nucleotides (TGNs). (A, left panel) 14-day experiment with once daily gavaging of vehicle, 6-thioguanine (6TG), 6-mercaptopurine (6MP), methylated 6TG (me6TG) or methylated 6MP (me6MP). Only 6TG 2.5 mg/kg/day caused SOS. N=6 C57Bl/6 male or female mice in each group. Box and whisker plots. Mann–Whitney, **p<0.01 6TG versus all other groups. (A, right panel) Representative liver histology from vehicle, 6TG, me6TG and me6MP livers. (B) Same experiment as (A). High-dose 6TG and 6MP but not me6TG or me6MP caused leucopenia. Statistics: Box and whisker plots of median, quartiles, range. Mann–Whitney, **p<0.01, ***p<0.001. (C, D). N=6 wild type (WT) versus hypoxanthine (guanine) phosphoribosyl transferase (Hprt)-deficient male and female mice gavaged once daily with 2.5 mg/kg/day 6TG for 10 days or with vehicle only or 0.5 mg/kg/day 6TG for 14 days. Hprt deficiency completely abrogated SOS (C) and significantly attenuated 6TG-induced leucopenia in the C57Bl/6 (D). There was a statistically significant higher leucocyte count in untreated control Hprt-deficient mice, which has not been reported previously. The mild leucopenia with 6TG in Hprt-deficient mice could have been due to an immunosuppressive effect of 6TG itself or other metabolites, see discussion. As expected 6TG caused significant leucopenia in WT animals, p=0.0004 for 6TG 0 vs 2.5 mg/kg/ day. Statistics: column graphs showing mean and SD of SOS (C) or leucocyte counts (D). Manne-Whitney, *p<0.05, **p<0.01. (E). Male C57Bl/6 mice were gavaged daily 2.5 mg/kg 6TG for 12 days, vehicle only, 0.5 mg/kg 6TG or 2.5 mg/kg 6MP for 30 days. TGNs accumulated in blood cells with 6TG and high-dose 6MP (Kruskal–Wallis, p=0.0137) but were not detected in the liver (data not shown).
Having found no evidence to support a role in the model for oral gavaged me6TG, me6MP or 6MP in SOS, we then investigated 6TG gavage in Hprt-deficient mice. These mice lack Hprt to convert 6TG to TGNs (figure 1). 6TG was gavaged daily for 10 days in C57Bl/6 and Hprt-deficient mice. Hprt-deficient mice did not lose weight (online supplementary figure 5), were protected from SOS in this experiment (figure 6C) and were also largely protected from immunosuppression (figure 6D). Only WT C57Bl/6 mice developed significant SOS and immunosuppression, which occurred as in previous experiments at the higher gavage dose of 2.5 mg/kg/day 6TG (figure 6C,D). These experiments demonstrate that 6TG immunosuppression and liver pathology are dependent on Hprt and therefore mediated by TGNs.
The degree of immunosuppression with thiopurine treatment is known to correlate with the accumulation of TGNs in red blood cells, which underpins current therapeutic drug monitoring, and white cells, the target of thiopurine therapy. We previously hypothesised that the liver metabolises thiopurine prodrugs to TGNs after which they may be transported by blood to other sites in the body,1 but it is not known if TGNs accumulate in hepatocytes as they do in red and white blood cells. To investigate, male C57Bl/6 mice were gavaged daily 2.5 mg/kg 6TG for 12 days, vehicle only, 0.5 mg/kg 6TG or 2.5 mg/kg 6MP for 30 days, and TGNs were measured in liver and peripheral blood after they were killed at the end of the gavage treatment. TGNs accumulated in the peripheral blood of treated animals (figure 6E), but were not detected in the liver of any of the treatment groups.
Inflammatory cells amplify 6TG-induced SOS
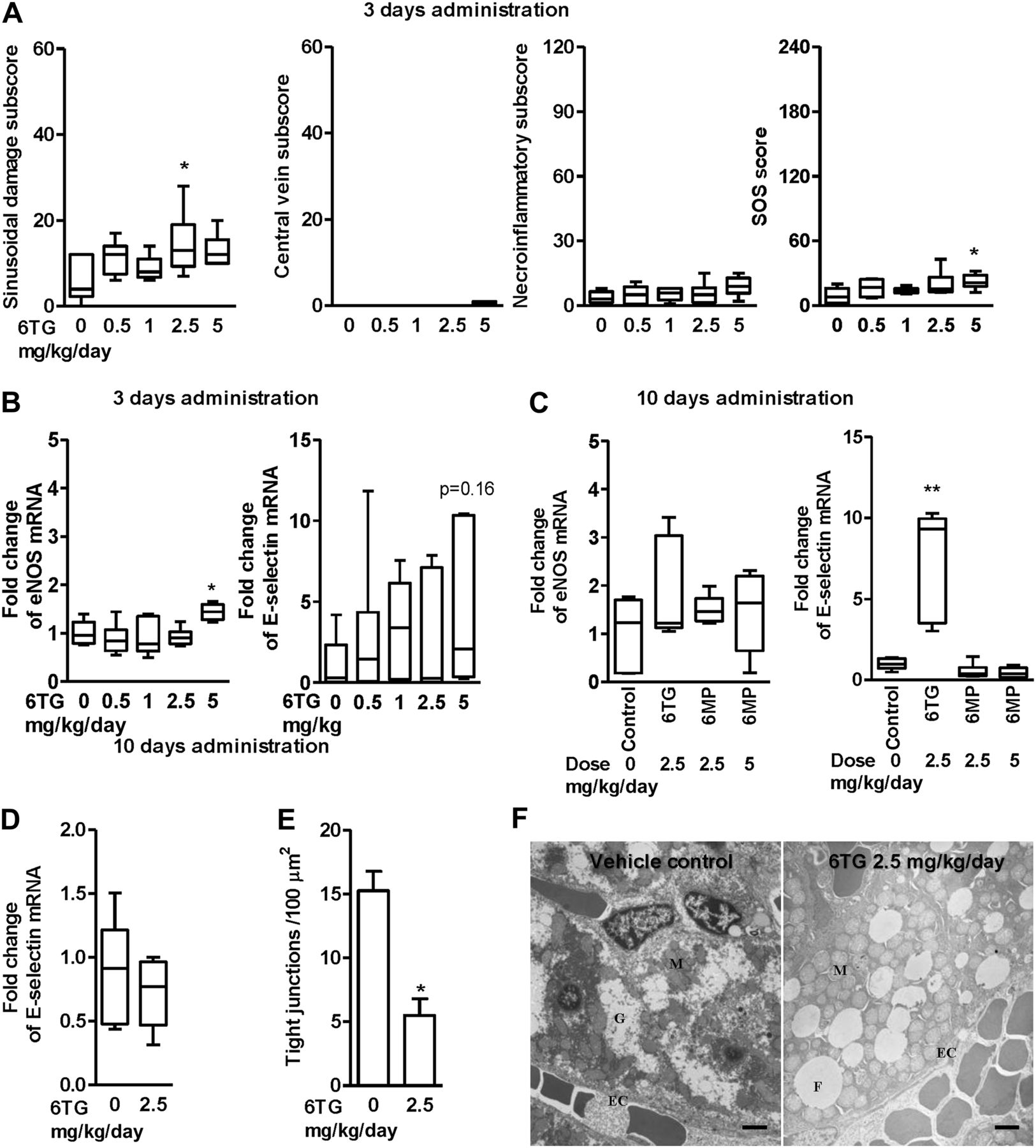
To identify the initiating events in the liver pathology, we examined livers from mice treated for 3 days with 6TG doses ranging from 0.5 to 5 mg/kg/day. SOS was barely discernable at 3 days with a 6TG dose of 5 mg/kg/day but the sinusoidal damage subscore trended upwards with increasing 6TG dose (figure 7A), consistent with sinusoidal endothelial damage being an early event as in the monocrotaline rat model of SOS. Furthermore, along with endothelial cell damage after 10–12 days' exposure to 6TG, a marked inflammatory cell infiltration was noted in the liver, as demonstrated by F4/80 staining of increased monocytes/macrophages, some of which appeared adherent to central vein endothelial cells (figure 4A). Therefore we investigated activation of endothelial cells. During the initial phase of an inflammatory response, stimuli such as histamine and thrombin cause endothelial cells to mobilise P-selectin from stores inside the cell to the cell surface,32 and in addition, endothelial nitric oxide synthase (eNOS) generates nitric oxide in blood vessels, which enhances leucocyte traffic in the small hepatic sinusoids, and a few hours later cytokines such as tumour necrosis factor-α induce E-selectin transcription and expression.33 While P-selectin promotes leucocyte adhesion and fast rolling on the endothelium in the absence of frank inflammation, E-selectin promotes slow rolling and leucocyte activation.34 We therefore investigated mRNA of eNOS and E-selectin but not P-selectin, which is constitutively transcribed by endothelial cells. The highest 6TG dose of 5 mg/kg/day upregulated eNos mRNA in the liver (figure 7B), but E-selectin mRNA was not significantly altered at 3 days. However by 9–12 days, E-selectin mRNA in the liver was upregulated 10-fold with gavaging 2.5 mg/kg/day 6TG but not with large doses of 6MP (figure 7C) and not with 6TG in Hprt-deficient mice (figure 7D). Ultrastructure at this same time point revealed fewer sinusoidal endothelial tight junctions (figure 7E), and swollen mitochondria and fat globules in hepatocytes in the 6TG-treated mice (figure 7F). Together these results suggest that 6TG treatment damages and activates hepatic endothelium, resulting in recruitment of inflammatory leucocytes such as monocytes and macrophages.

6-thioguanine (6TG)-induced sinusoidal obstructive syndrome (SOS) is associated with endothelial activation and loss of fenestrated tight junctions. (A) To study early events, n=6 wild type (WT) mice were gavaged once daily with 6TG 0–5 mg/kg/day for 3 days. Frank SOS was barely evident in WT mice at 3 days with 6TG 2.5–5 mg/kg/day; the sinusoidal damage subscore appeared most altered. (B) Fold change at 3 days in hepatic endothelial nitric oxide synthase (eNos) and E-selectin mRNA from whole liver (mRNA was normalised to VE-cadherin mRNA, see online supplementary figure 1). There was upregulation of eNos but not E-selectin mRNA with the highest 6TG dose. (C, D) N=6 animals in each group. Once daily gavage of 6TG 2.5 mg/kg/day for 10 days caused a 10-fold increase in E-selectin (but not eNos) mRNA. This did not occur with 6-mercaptopurine (6MP) (C) or in hypoxanthine (guanine) phosphoribosyl transferase (Hprt)-deficient mice (D) (eNos mRNA data are not shown for Hprt-deficient mice). (E, F) Transmission electron microscopy (TEM) of livers from 10-day 6TG 2.5 mg/kg/day and 14-day vehicle-gavaged mice. (E) The number of intact endothelial cell (EC) tight junctions was reduced with 6TG 2.5 mg/kg/day (N=4). (F) Representative TEM. There was fat globule (F) accumulation, altered mitochondria (M) and absent glycogen (G) in hepatocytes with 6TG treatment. Scale bars 2 μm. Statistics: test versus control, Mann–Whitney, *p<0.05, **p<0.01.
The importance of inflammatory cell recruitment in amplifying SOS in the model was assessed by gavaging 6TG in P-selectin/E-selectin double knock-out (PE-selectin −/−) mice on a C57Bl/6 background.35 These mice lack expression of P-selectin and E-selectin on the surface of endothelial cells and therefore the cascade of leucocyte adhesion, rolling and diapedesis is impaired. In addition, absence of E-selectin may reduce activation of infiltrating immune cells.36 6TG in PE-selectin −/− mice resulted in a markedly reduced infiltration of macrophages into liver parenchyma (figure 8A) and a reduced SOS score (figure 8C). The near complete (>80%) abrogation of SOS in PE-selectin −/− mice was due to a reduction in all the SOS subscores. Reduction in inflammatory cells in the livers of PE-selectin −/− mice accounted for <25% of the necroinflammatory subscore because other elements of this subscore, specifically apoptosis and steatosis, accounted for >75% of the reduction in the necroinflammatory subscore. The slightly elevated SOS score in the 6TG-gavaged PE-selectin −/− mice was principally due to sinusoidal endothelial damage. Figure 8B shows that leucocyte counts were reduced by high-dose 6TG in the PE-selectin −/− mice, demonstrating that while SOS was attenuated by inhibition of P-selectin and E-selectin, 6TG still induced immunosuppression.

6-thioguanine (6TG)-induced sinusoidal obstructive syndrome (SOS) is attenuated by blocking endothelial cell activation. 6TG 2.5 mg/kg or vehicle was gavaged once daily for a median of 11 days in PE-selectin −/− and wild type (WT) mice. (A) Representative immunohistochemistry with F4/80 antibody, scale bar 100 μm. 6TG was associated with a prominent infiltrate of macrophages in the liver of 6TG-treated WT mice but not PE-selectin −/− mice. (B) Leukocyte counts were higher in venous blood of vehicle-control PE-selectin −/− mice, which was expected due to the reduced margination of leucocytes.36 6TG was immunosuppressive in the double-knockout and WT C57Bl/6 mice. (C) SOS scoring demonstrated marked attenuation of SOS in PE-selectin −/− mice. This was mainly due to marked reductions in the central vein damage and necroinflammatory subscores. Statistics: Mann–Whitney, *p<0.05, **p<0.01. Not shown in the figure, p values <0.001 for all scores in WT animals with 6TG versus vehicle treatment.
Splitting the 6TG dose significantly reduces SOS and colitis
Based on the Hprt-deficient animal experiments and the absence of SOS with daily low-dose 6TG, we hypothesised that the initial insult initiating SOS is caused by a high maximum concentration (Cmax) of TGNs in the liver and that consequently SOS can be avoided by reducing the concentration of 6TG in the portal-hepatic circulation. This presumably occurs naturally with therapeutic dosing of 6MP or AZA prodrugs, which require multiple enzymatic steps in liver, erythrocytes and leucocytes to generate TGNs.1 This hypothesis was tested in normal mice by splitting the daily dose of 2.5 mg/kg/day 6TG over 11–12 days. Figure 9A illustrates that 1.25 mg twice daily gavages of 6TG significantly reduced SOS (p=0.03 compared with littermates gavaged 2.5 mg/kg/day 6TG in the morning and vehicle control in the afternoon).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Split dosing of 6-thioguanine (6TG) ameliorates sinusoidal obstructive syndrome (SOS) and colitis. (A) Female C57Bl/6 mice were gavaged twice daily in a 14-day experiment with three groups: 1.25 mg/kg 6TG (total 2.5 mg/kg/day, n=7), vehicle control (n=13) or 2.5 mg/kg/day 6TG in the morning and vehicle gavage in the afternoon (n=12). Subscores and total SOS score were all decreased by split dosing of 6TG 2.5 mg/kg/day. Statistics: box and whisker plots of median, quartiles, range. (B) n=6 male or female Winnie mice were gavaged twice daily with either 1.25 mg/kg 6TG (total 2.5 mg/kg/day) or vehicle control, or 2.5 mg/kg/day 6TG in the morning and vehicle gavage in the afternoon for 11 days. Histological colitis scores from proximal colon (PC), mid-colon (MC) and distal colon (DC) showed significantly improved spontaneous colitis in Winnie mice24 ,37 treated with 6TG 1.25 mg/kg twice daily or 2.5 mg/day gavaged once daily. There was a trend for increased treatment efficacy with once daily gavage (p=0.07). (B, right panel) Representative haematoxylin and eosin (H&E) histology from the distal colon of a split-dose treated Winnie mouse shows improvement in the colitis; scale bars represent 100 μm. (C, D) Venous blood leucocyte counts at killing from the animal experiments shown in figure 9A,B respectively. Split dosing 1.25 mg/kg/day caused immunosuppression, which trended less than immunosupression in the 2.5 mg/kg/day treated groups (p=0.08). Other statistics: Mann–Whitney, *p<0.05, **p<0.01 comparisons with vehicle controls.
The same protocol was tested in the C57Bl/6 Muc2Winnie/Winnie (Winnie) model of spontaneous colitis (figure 9B). The spontaneous colitis in Winnie resembles ulcerative colitis and results from a single nucleotide polymorphism in Muc2 that causes misfolding of Muc2 protein, endoplasmic reticulum stress in intestinal epithelium and a T helper 17 predominant inflammatory response.24 ,37 Split dosing in Winnie resulted in a significant improvement in the colitis (figure 9B), which was not unexpected because 6TG should dampen the inflammatory response, as indicated by parallel leucopenia (figure 9D). Split dosing in WT C57Bl/6 mice caused a moderate immunosuppression which trended less than single daily dosing of 6TG (figure 9C). But while SOS parallelled leucopenia in the single daily gavage experiments, in this experiment split-dosing caused uncoupling of SOS from immunosuppression.
Discussion
We have developed a novel murine model of SOS caused in a dose-dependent manner by orally administered 6TG. Damage was mediated by TGNs, and was reduced by inhibition of leucocyte recruitment by the activated hepatic endothelium. While a previous study of C57Bl/6 mice treated with 6TG did not detect SOS-VOD,27 we believe the explanation for this difference is straightforward; 6TG was not gavaged in that study. Rather, it was given ad libitum in the drinking water, which would result in lower concentrations of active drug in the portal and hepatic circulation of the mice. Our model provides important insights into the aetiology of 6TG-induced SOS-VOD, and suggests that simple changes to administration can avoid this dangerous ADR.
6TG dose and SOS
There is controversy in the IBD literature about whether 6TG-SOS is idiosyncratic or dose related, but the findings of our model are clearly consistent with a dose-related phenomenon. The belief that 6TG caused an idiosyncratic NRH was based mainly on one retrospective study of 111 patients in whom 6TG doses were not reported, and out of which 26 underwent liver biopsy and 18 were reported to have NRH. On the other side of the controversy38 Dutch gastroenterologists reported on a prospective series of 106 patients with IBD prescribed low-dose 6TG (0.3 mg/kg/day) for a median duration >2 years, all of whom underwent liver biopsy at 6 months.39 Apart from noting occasional sinusoidal dilatation—a histological feature in the sinusoidal damage subscore found with vehicle or low 6TG doses (figure 5B)—no NRH or VOD was detected in their prospective series with controlled dosing.
While frank SOS-VOD/NRH is occasionally reported in patients treated with AZA or 6MP40 ,41 the prevalence does not appear to be above the background prevalence in the general population.18 Furthermore it is clear from the leukaemia literature that VOD occurs with increasing dose exposure to 6TG but not with 6MP.16 ,42 The robust incidence data from the Children's Oncology Group CCG-1952 clinical trial reported 28% VOD in the children receiving 60 mg 6TG/m2/day, 22% VOD in children receiving 50 mg 6TG/m2/day, but no cases of VOD in the children receiving 75 mg 6MP/m2/day without 6TG.16 Most of the children in this large randomised controlled trial were aged between 2 and 5 years, from which it can be adduced that the daily maximum 6TG and 6MP doses were equivalent to about 5 and 6 mg/kg respectively. These paediatric doses translate to about 2.5 and 3 mg/kg of 6TG and 6MP in an adult,43 which are comparable to the higher doses used in our experimental model.
SOS and immunosuppression are due to TGNs
Mice with Hprt deficiency had normal body weight and no SOS after high-dose 6TG, which was in sharp contrast to WT mice. Hprt deficiency also largely prevented the immunosuppression, which is responsible for leucopenia. This was expected because it is the TGNs, in particular TGTP, which compete for intracellular GTP-binding RAS proteins in leucocytes2 and intercalate in DNA.44 ,45 The mild immunosuppression noted with high-dose 6TG in the Hprt-deficient mice could have resulted from other mechanisms, including from bacterial metabolism. We also demonstrated that gavage of supra-pharmacological doses of me6TG caused no immunosuppression (or SOS). However, methylation of high-dose 6TG can be predicted to deplete the folate-dependent cofactor of thiopurine S-methyltransferase (TPMT), S-adenosyl-methionine. Hence the mild immunosuppression in Hprt-deficient mice could have resulted from either S-adenosyl-methionine or folate starvation of leucocytes.46
TGNs, like all nucleotides, survive solely as intracellular metabolites.1 It follows that 6TG-induced SOS results from TGNs (thioguanosine monophosphate, thioguanosine diphosphate, TGTP or a TGN metabolite such as a methylTGN) produced directly within liver cells (reticuloendothelial cells, hepatocytes), which produce a cytotoxic effect that is restricted to the sinusoids and adjacent tissue where the concentration of 6TG is highest. The necessity for metabolic conversion and intracellular accumulation of TGN may explain the slower action of 6TG compared with the cytotoxicity of monocrotaline, which acts on the endothelial cells by formation of protein-binding free radicals to cause SOS within 12–24 h.21
In humans, the bioavailability of oral thiopurines is variable.47–49 Much of the absorbed prodrug is catabolised by first pass methylation and deamination/oxidation in the small intestine and liver (figure 1).47 ,50 ,51 However, some TGNs are produced by liver cells that are utilising the purine synthesis pathway to generate GTP. It can be inferred from the SOS scores that the level of TGNs in liver cells is correlated to the maximum concentration of 6TG in the portal circulation. Furthermore, SOS did not occur with 6TG ≤0.5 mg/kg/day over 28 days and the SOS score was not increased with longer gavaging of 6TG 0.5 mg/kg/day from 14 to 28 days. This together with the inability to detect TGNs in the liver, suggests that the toxicity from TGN metabolites in the liver is not cumulative. We propose that intracellular TGNs do not accumulate in hepatocytes, which is in contrast to activated lymphocytes or erythrocytes that are mainly exposed to lower concentrations of 6TG. This can explain why chronic low-dose 6TG (median >30 months, 0.3 mg/kg/day) was found to be safe38 while still effecting some immunosuppression. AZA and 6MP do not cause SOS because their metabolism requires other enzymatic steps in hepatocytes including that of rate-limiting inosinemonophosphate dehydrogenase,52 so that intracellular TGNs always remain low in hepatocytes with AZA or 6MP treatment.1 The faster immunosuppressive action of 6TG versus 6MP in our experiments can be explained if TGNs accumulate faster in leucocytes with 6TG administration compared with administration of AZA or 6MP. This is consistent with the known pharmacokinetics of oral thiopurines in humans, in whom a steady-state intracellular concentration of TGNs is reached within 4 weeks with 6TG,53 but requires longer for 6MP.54
Inhibition of endothelial cell activation attenuates SOS
We have shown that sinusoidal damage was a prominent feature of this model, which is also the case with 6TG and VOD/NRH in humans. The hepatic sinusoidal endothelial cells are exposed to the highest concentration of oral gavaged 6TG. This suggests the following sequence of events: hepatic metabolism of portal 6TG results in intracellular TGNs which cause off-target damage to liver cells, including endothelial cells, which then upregulate E-selectin. Cell surface expression of P-selectin and E-selectin on activated endothelial cells results in enhanced recruitment of inflammatory leucocytes,33 which is reversed by deletion of P-selectin and E-selectin genes, preventing the cascade of leucocyte adhesion and diapedesis into the damaged tissue, to markedly attenuate SOS in the model.
There are many similarities between the SOS caused by monocrotaline in the rat model and our model—sinusoidal congestion, central vein endothelial damage, necroinflammatory infiltrate—but there are also differences. In particular, sinusoidal dilatation, which is also a feature of the earliest 6TG hepatotoxicity in humans,39 was not reported in the monocrotaline Sprague-Dawley rat model.22 Also monocrotaline causes SOS in the rat model within 12–24 h21 whereas 6TG's toxic effect even with the highest dose of 5 mg/kg/day at 3 days failed to manifest as frank SOS (figure 7A), which we have demonstrated relates to the fact that 6TG must first be converted to TGNs (figure 6C). It is also important to emphasise that both models are acute. Neither completely reproduces the clinicopathological entity of VOD, which goes on to manifest with clinical portal hypertension, or the pathology of NRH, which is associated with regenerative liver nodules in addition to portal hypertension. In general, the C57Bl/6 mice appeared to be quite sensitive to 6TG, with most mice becoming pancytopenic with doses of 6TG ≥1 mg/kg/day. One possible explanation for this sensitivity compared with humans is that the inbred C57Bl/6 mice have lower TPMT (unpublished data).55
Splitting the 6TG dose uncouples immunosuppression from SOS
SOS is caused by TGNs in the liver compartment but immunosuppression is mainly caused by TGNs in the blood compartment, and consequently once daily gavage of 6TG resulted in immunosuppression, which parallelled SOS and, in Winnie, improvement of colitis (figure 9). However, while immunosuppression parallelled the improvement in colitis, SOS did not parallel immunosuppression or the improvement in colitis with split dosing. Split dosing delivers a lower concentration of 6TG to the portal circulation, which results in less intracellular TGNs in the liver. The reason for the trend for greater immunosuppression and the parallel greater improvement of colitis with once daily dosing in Winnie is likely related to the bioavailability of TGNs because first pass catabolic and anabolic metabolism will be different with the two modes of delivering the same daily dose of 6TG.
There are clinically important implications that arise from the results of the model. IBD is a worldwide disease. There is a clinical need for an economical faster-acting, more tolerated immunomodulatory therapy. We hypothesised that the initial insult to cause SOS is related to Cmax of 6TG in the hepatic sinusoids and showed using the Hprt-deficient mice that SOS could be avoided by preventing the accumulation of TGNs in the liver. We found support for this hypothesis with the split dose experiment in which it can be inferred that Cmax of 6TG is reduced (figure 9A). We concluded that this prevented SOS but that it did not prevent immunosuppression due to accumulation of TGNs in leucocytes. We also demonstrated a rapid response with split dosing and once daily gavage dosing in abrogating the spontaneous colitis in the Winnie mouse model (figure 9B), and that this therapeutic effect parallelled the degree of immunosuppression. The result of the split-dose experiment in the model suggests that there would be clinical advantage in investigating in humans a mode of drug delivery which reduced Cmax of 6TG in the portal circulation and thence TGNs in the liver. Experiments are currently being undertaken by our research group to ascertain hepatoportal vein and peripheral blood pharmacokinetics of novel experimental 6TG delivery formulations. We also demonstrated that inhibition of P-selectin and E-selectin expression can reduce SOS by abrogating the influx and activation of inflammatory cells into the liver. This suggests the possibility of another clinical avenue, which could lead to the prevention of SOS-VOD due to 6TG and other drugs that cause SOS-VOD. Taken together, the novel findings are clinically relevant to 6TG administration and the prevention of SOS.
Acknowledgments
We thank Rachel Adams, Debra Roche, Thu Tran and Toshna Singh for expert technical assistance, and Dr Sumaira Hasnain for careful reading of the manuscript. IW is in receipt of an NHMRC Career Development Research Fellowship, MAM is a NHMRC Senior Research Fellow, THJF is an NHMRC Clinical Practitioner Fellow, J-PL was a Cancer Council Queensland Research Fellow. Iulia Oancea was funded in part by the Australian Crohn's and Colitis Association.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement 1 - Online Supplementary Figure 1
Footnotes
Correction notice This article has been corrected since it was published Online First. The author name Ingrid Winkler has been amended to Ingrid G Winkler.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.