Article Text

Abstract
Objective Mutations in the nucleotide-binding oligomerisation domain-containing protein 2 (NOD2) gene remain the strongest genetic determinants for Crohn's disease (CD). Having previously identified vimentin as a novel NOD2-interacting protein, the authors aimed to investigate the regulatory effects of vimentin on NOD2 function and the association of variants in Vim with CD susceptibility.
Design Coimmunoprecipitation, fluorescent microscopy and fractionation were used to confirm the interaction between NOD2 and vimentin. HEK293 cells stably expressing wild-type NOD2 or a NOD2 frameshift variant (L1007fs) and SW480 colonic epithelial cells were used alongside the vimentin inhibitor, withaferin A (WFA), to assess effects on NOD2 function using the nuclear factor-kappaB (NF-κB) reporter gene, green fluorescent protein-LC3-based autophagy, and bacterial gentamicin protection assays. International genome-wide association meta-analysis data were used to test for associations of single-nucleotide polymorphisms in Vim with CD susceptibility.
Results The leucine-rich repeat domain of NOD2 contained the elements required for vimentin binding; CD-associated polymorphisms disrupted this interaction. NOD2 and vimentin colocalised at the cell plasma membrane, and cytosolic mislocalisation of the L1007fs and R702W variants correlated with an inability to interact with vimentin. Use of WFA demonstrated that vimentin was required for NOD2-dependent NF-κB activation and muramyl dipeptide-induced autophagy induction, and that NOD2 and vimentin regulated the invasion and survival properties of a CD-associated adherent-invasive Escherichia coli strain. Genetic analysis revealed an association signal across the haplotype block containing Vim.
Conclusion Vimentin is an important regulator of NOD2 function and a potential novel therapeutic target in the treatment of CD. In addition, Vim is a candidate susceptibility gene for CD, supporting the functional data.
- Inflammatory bowel disease
- Crohn's disease
- NOD2
- vimentin
- E coli
- autophagy
- genetic association studies
- IBD
- bacterial pathogenesis
- cell signalling
- paediatric gastroenterology
- immunology
- IBD basic research
- E coli and IBD
- enteric bacterial microflora
- bacterial pathogenesis
- IBD models
- genetics
- immunology
Statistics from Altmetric.com
- Inflammatory bowel disease
- Crohn's disease
- NOD2
- vimentin
- E coli
- autophagy
- genetic association studies
- IBD
- bacterial pathogenesis
- cell signalling
- paediatric gastroenterology
- immunology
- IBD basic research
- E coli and IBD
- enteric bacterial microflora
- bacterial pathogenesis
- IBD models
- genetics
- immunology
Significance of this study
What is already known on this subject?
-
Evidence suggests that Crohn's disease (CD) arises from a defective innate immune response to enteric bacteria.
-
Mutations in the nucleotide oligomerisation domain protein 2 (NOD2) gene remain the strongest single genetic determinant of CD.
-
Several lines of research have demonstrated that the intermediate filament vimentin, is involved in bacterial pathogenicity.
-
Escherichia coli strains with an adherent and invasive phenotype (AIEC) have been consistently isolated by independent investigators from patients with CD and ileal disease.
What are the new findings?
-
The leucine-rich repeat domain of NOD2 contains the functional elements required for the binding of vimentin, with CD-associated polymorphisms in this domain disrupting the interaction.
-
NOD2 and vimentin colocalise at the cell plasma membrane, and mislocalisation of the L1007fs and R702W NOD2 variants to the cytosol correlates with an inability to interact with vimentin.
-
Vimentin regulates NOD2 activities that are dependent on its plasma membrane localisation, such as nuclear factor-kappaB (NF-κB) activation, autophagy and handling of CD-associated AIEC. The gene encoding vimentin (Vim) is a novel candidate susceptibility gene for CD.
How might it impact on clinical practice in the foreseeable future?
-
This study describes a novel functional link between NOD2, vimentin and CD-associated E.coli identifying vimentin as a potential molecular target for the treatment of CD.
-
The vimentin inhibitor, withaferin A, a steroidal lactone from the plant Withania somnifera, has potential for the treatment of CD as either a prophylactic or therapeutic modality.
Introduction
Crohn's disease (CD), a major form of inflammatory bowel disease (IBD), is a chronic disease of the intestinal tract associated with significant morbidity. To date, a substantial volume of work on CD pathogenesis suggests that the disease results from a dysregulated immune response to enteric bacteria in a genetically susceptible host.1 Major CD susceptibility pathways implicated through recent genome-wide association scanning (GWAS) include the innate immune response (NOD2), the more specific, acquired T cell response (IL23R) and autophagy (ATG16L1, IRGM).2 ,3 Examination of the disease-associated microbiome has also implicated several potentially causative agents, most notably Escherichia coli strains with an adherent and invasive phenotype (AIEC), which have been consistently isolated by independent investigators from patients with CD and ileal disease.4
Nucleotide oligomerisation domain protein 2 (NOD2) is a receptor for the bacterial cell wall component, muramyl dipeptide (MDP).5 Although originally characterised as a cytosolic receptor, recent studies have demonstrated that NOD2 is localised to, and activated at, the cell plasma membrane.6 ,7 Mutations in NOD2 remain the strongest single genetic determinant for CD, with three disease-associated polymorphisms, namely Arg702Trp (R702W), Gly908Arg (G908R) and Leu1007fsinsCys (L1007fs), having been shown to affect the leucine-rich repeat (LRR) domain.8 Although carriage of two of the CD-associated NOD2 polymorphisms confers a 20–40-fold risk of developing CD, the mechanisms whereby mutations in this gene influence disease pathogenesis are still unclear.
To date only a small number of NOD2-interacting proteins have been identified,9 and currently there is limited knowledge about their functional effect on NOD2. In our previous yeast-two-hybrid (Y2H) screen, we identified a novel interaction between NOD2 and the cytoskeletal protein, vimentin.10 Vimentin is the major intermediate filament (IF) protein present primarily in mesenchymal cells, with several lines of research having recently demonstrated its role in bacterial pathogenicity.11 ,12 Here we aimed to characterise this interaction in mammalian cells and to determine how vimentin affects NOD2 function. In addition, we assessed the association signal of the single-nucleotide polymorphisms (SNPs) in the gene encoding vimentin (Vim) with regard to the genetic susceptibility to CD. We report that vimentin interacts with NOD2 at the cell plasma membrane through the LRR domain, with CD-associated polymorphisms in NOD2 abrogating this interaction. We also demonstrate that vimentin regulates NOD2 activities that are dependent on its plasma membrane localisation (such as nuclear factor-kappa B (NF-κB) activation, autophagy and bacterial handling) as well as showing that Vim is a candidate susceptibility gene for CD.
Methods
Plasmids
The generation of haemagglutinin-tagged (HA) NOD2 variants (R702W, G908R and L1007fs) have been described previously.10 For the generation of HA-NOD2 (1–693), lacking the C-terminal LRR domain, and HA-NOD2 (1–247), comprising only the two N-terminal caspase activation and recruitment domains (CARDs), site-directed mutagenesis (Stratagene, San Diego, California, USA) was used to introduce a stop codon at amino acids 694 or 248. All primer sequences are available on request. NF-κB-luciferase reporter plasmids were a gift from Dr Lesley Stark (University of Edinburgh, Edinburgh, UK).
Drug treatments
Withaferin A (WFA) and dimethyl sulphoxide (DMSO) were purchased from Sigma (Dorset, UK). WFA stock solution (10 mg/ml in DMSO) was diluted in Dulbecco's modified Eagle medium (DMEM) to working concentrations between 2 and 10 μM. Equivalent amounts of DMSO only were used as negative control for WFA. L18-MDP was purchased from InvivoGen (San Diego, California, USA). L18-MDP stock solution (10 mg/ml in water) was diluted in DMEM to a working concentration of 1 or 50 μg/ml. Tumour necrosis factor alpha (TNFα) was purchased from PeproTech (Rocky Hill, New Jersey, USA). TNFα stock solution (5 μg/ml in water) was diluted in DMEM to a working concentration of 50 ng/ml.
Antibodies
Vimentin, light chain 3 (LC3), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and histone H1 antibodies were purchased from Abcam (Cambridge, UK), E-cadherin antibody from BD Biosciences (San Diego, California, USA), NOD2 antibody from Cayman Chemical (Ann Arbor, Michigan, USA), and HA-11 antibody from Covance (Emeryville, California, USA). β-Actin and P65/RelA antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA).
Y2H spotting assay
Yeast strain AH109 was cotransformed with a plasmid containing the full-length vimentin cDNA, wild-type NOD2 or CD-associated mutant NOD2 R702W using a small-scale LiAc transformation procedure (Yeast Protocols Handbook PT3024-1; Clontech, Santa Clara, California, USA). Cotransformants were selected on SD/Leu−Trp− plates and spotted on to selective medium, SD/His−Leu−Trp−.
Cell culture, transfection and immunoblotting
HEK293 cells were grown in DMEM, and SW480 cells in Leibovitz's medium supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, California, USA). For Lipofectamine 2000 transfection, 2 μl Lipofectamine (Invitrogen) was used for every 1 μg DNA. Cells were harvested after 16–18 h and lysed in ice-cold extraction buffer (50 mM Tris (pH 7.6), 150 mM NaCl2, 5 mM EDTA, 0.5% Nonidet P40, 5 mM NaF, 1 mM sodium vanadate, 1 × Protease Inhibitor Cocktail) for 30 min and centrifuged. Protein content of cell extracts was measured using Bio-Rad reagent (Bio-Rad, Hemel Hempstead, UK). Samples were resolved by denaturing gel electrophoresis, typically 4–12% Novex precast gels (Invitrogen), electrotransferred to Hybond C-extra nitrocellulose membrane (Amersham, Little Chalfont, UK), and incubated with primary antibody overnight at 4°C. After being washed, blots were incubated with secondary antibody, either horseradish peroxidase-conjugated anti-rabbit or anti-mouse (Dako, Cambridgeshire, UK), for 1 h at room temperature. Proteins were visualised by incubation in an ECL western blotting analysis system (Amersham) or with Immobilon Western Chemiluminescent HRP Substrate (Millipore, Durham, UK).
Generation of stable cell lines
NOD2 or NOD2-L1007fs cells were sub-cloned into the p3XFLAG-myc-CMV-26 expression vector (Sigma, Gillingham, Dorset, UK). HEK293 cells were transfected with FLAG-EMPTY, FLAG-NOD2 or FLAG-NOD2-L1007fs vectors using Lipofectamine 2000. After 48 h of transfection, cells were cultured in DMEM supplemented with 800 μg/ml geneticin (Invitrogen), and resistant colonies were picked and screened for NOD2 expression using NOD2-specific antibodies.
Immunoprecipitation
For immunoprecipitation of endogenous vimentin or exogenous HA-NOD2, appropriate antibody bound to protein G beads (Amersham) was incubated overnight at 4°C with cell extract (∼1 mg) diluted to a volume of 500 μl in extraction buffer. Bead pellets were then washed before being resuspended in 3×SDS-loading buffer, and associated proteins were analysed by denaturing gel electrophoresis and immunoblotting.
Cell fractionation
For subcellular fractionation of HA-NOD2-expressing cells, the Qproteome Cell Compartment Kit (Qiagen, Valencia, California, USA) was used according to the manufacturer's protocol.
Immunofluorescence microscopy
Cells were seeded on 19 mm borosilicate glass cover slips at a density of 2×104 cells. After 24 h, if required, cells were transfected with HA-NOD2 DNA constructs for a further 16 h. Cells were then fixed with 4% paraformaldehyde, permeabilised with phosphate-buffered saline (PBS)/0.1% Triton X-100 and blocked with PBS containing 10% fetal calf serum. For protein detection, primary antibodies were incubated overnight at 4°C. Cells were then incubated for 1 h at room temperature with fluorescein isothiocyanate- or tetramethyl rhodamine isothiocyanate-conjugated anti-mouse or anti-rabbit secondary antibodies (Invitrogen). Where appropriate, cells were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI). Images were captured using a Carl Zeiss Axioskop 2 fluorescent microscope and analysed using Image J software (National Institutes of Health, Bethesda, Maryland, USA).
Luciferase assay
Dual-luciferase reporter assays (Promega, Madison, Wisconsin, USA) were performed according to the manufacturer's protocol. Briefly, SW480 cells were seeded on 24-well plates for 24 h before transfection with NF-κB-luciferase wild-type (NF-κB-luc-WT) or mutant with NF-κB-binding sites deleted (NF-κB-luc-Mut) (both 200 ng) and a control for transfection efficiency (Renilla, 100 ng). After 16 h, cells were treated for 6 h with L18-MDP (1 μg/ml) or for 2 h with TNFα (50 ng/ml) in the absence or presence of various concentrations of WFA (2 h) or DMSO control (2 h) and harvested for luciferase assays.
Gentamicin protection assay
For invasion assays, cells were infected with the E coli strains isolated from the ileum of a patient with CD (CUICD541-10) or healthy ileum (CUH17-1) at a multiplicity of infection of 10 for 2 h. After the infection period, cells were washed and incubated for a further 2 h in medium containing 100 μg/ml gentamicin to kill extracellular bacteria; cells were then lysed for 10 min. Lysates were serially diluted and plated on Luria-Bertani (LB) agar plates; colonies were enumerated after overnight incubation at 37°C. Where appropriate, cells were preincubated with WFA or appropriate antibodies for 30 min before infection. For survival assays, the procedure was performed as above except that, after incubation in 100 μg/ml gentamicin, cells were incubated for a further 24 h in medium containing 20 μg/ml gentamicin. Where appropriate, cells were treated with WFA after infection.
Analysis of gene-wide susceptibility signal for Vim using an international GWAS meta-analysis
As members of the UK IBD Genetics Consortium and the larger International IBD Genetics consortium (IIBDGC online supplementary file), we now have access to novel genetic data generated from large case–control studies. Although a meta-analysis of CD GWAS data has previously been published,2 we were able to interrogate a new improved meta-analysis of GWAS data imputed with the 1000 genomes reference set (http://www.1000genomes.org). To assess variants in Vim (the gene encoding vimentin on chromosome 10 between positions 17 270 258 and 17 279 592 [Ensembl Release 65 - ENSG00000026025]; Build 37) for association with CD susceptibility, we obtained SNP-specific p values generated from these analyses of seven individual CD datasets encompassing 5956 patients with CD and 14 927 healthy controls (http://www.broadinstitute.org/mpg/ricopili/). p Values were generated with principal component analysis as covariates, therefore correcting the whole analysis for population stratification and interstudy differences. Haploview software version 4.2 (http://www.broad.mit.edu/mpg/haploview)13 was then used to visualise the haplotype structure (using solid spine of linkage disequilibrium) surrounding Vim to determine the areas of strongest signal.
Statistical analysis
For the functional assays, scanning densitometry was performed using Scion Image Software (National Institutes of Health). Results are reported as the mean±SD assuming normally distributed variables, with p values calculated using an unpaired t test in GraphPad V.4.03 (GraphPad Software). R V.2.14.1 was used to generate the GWAS SNP scatterplots using the ggplot2 package (http://http://had.co.nz/ggplot2/).
Results
Vimentin is a NOD2-interacting protein in mammalian cells
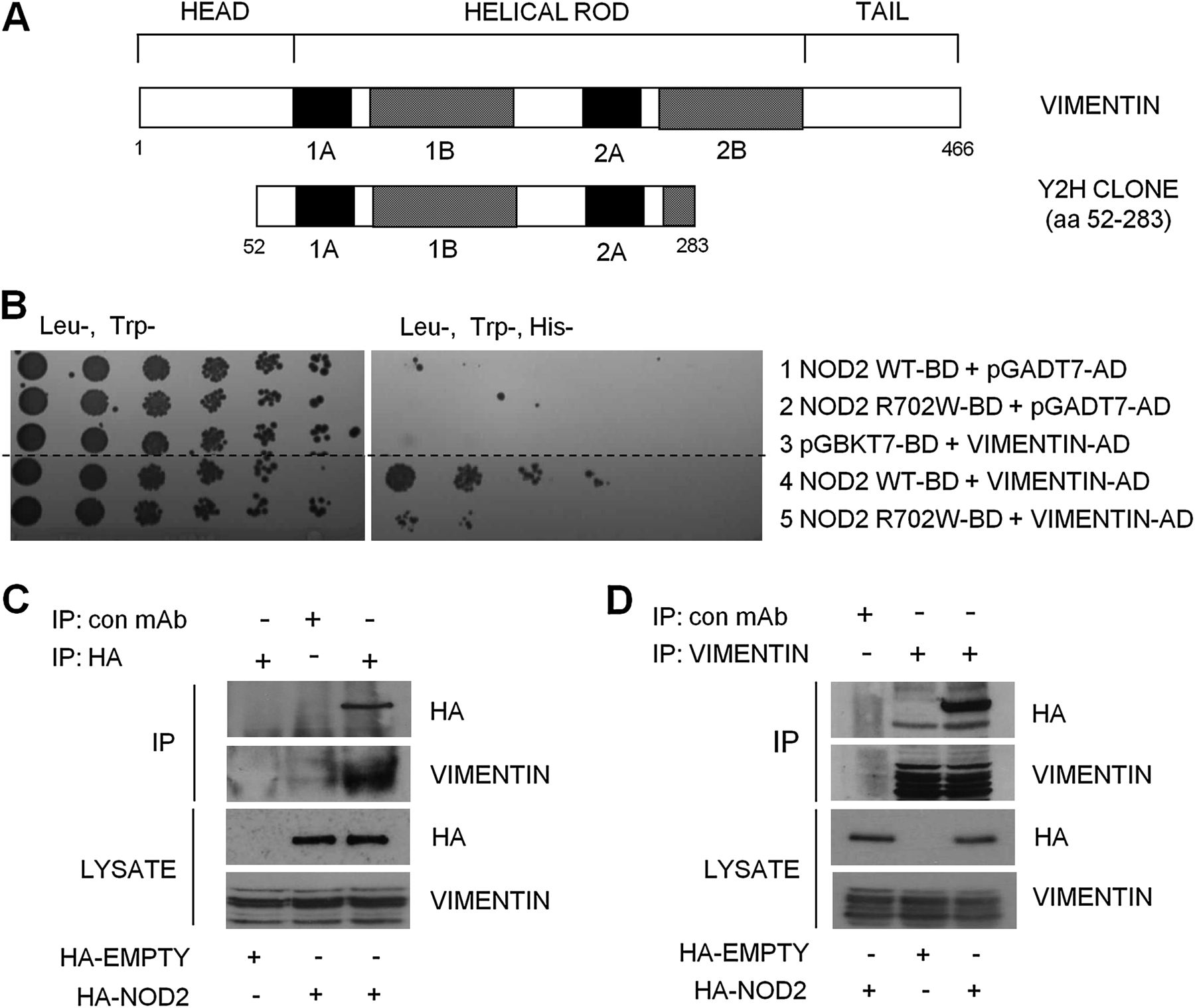
One clone identified by the Y2H screen10 as a novel NOD2 interactor corresponded to amino acids 52–283 of the IF protein, vimentin (figure 1A). To confirm this interaction, plasmids containing a full-length vimentin open reading frame fused to an activation domain were transformed into yeast along with full-length NOD2 or NOD2 with the inactivating polymorphism (R702W). NOD2 interacted with vimentin to activate the reporter gene, His3, whereas NOD2-R702W interacted less well with vimentin resulting in reduced activation (figure 1B). No activation of the reporter gene was observed when vimentin, NOD2 or NOD2-R702W alone was transformed into yeast (figure 1B). To confirm the NOD2–vimentin interaction in a cellular context, we transfected cells with HA-NOD2 and lysates immunoprecipitated with HA-specific antibodies, vimentin-specific antibodies or non-specific antibody as control. Results show that NOD2 and vimentin form a stable protein complex in cell lysates, since the immunoprecipitation of NOD2 enriched vimentin protein (figure 1C), and conversely the immunoprecipitation of vimentin enriched NOD2 protein (figure 1D). These results support the Y2H data and demonstrate that vimentin interacts with NOD2 in mammalian cells.

Identification of vimentin as a NOD2-interacting protein. (A) Schematic showing the structure of vimentin and the vimentin clone identified as binding to NOD2 in our yeast-two-hybrid assay. (B) Yeast strain AH109 was cotransformed with a plasmid containing the yeast expression vector pGADT7-AD, full-length vimentin cDNA, wild-type NOD2 or Crohn's disease (CD)-associated variant NOD2-R702W as shown. Cotransformants were selected on Leu−Trp−)plates and spotted on to selective medium (His−Leu−Trp−). (Dashed line indicates where irrelevant lanes have been removed.) (C) SW480 cells were transfected with haemagglutinin (HA)-NOD2 or HA-empty vector and extracts immunoprecipitated with HA-11-specific antibodies or non-specific antibody as control. Precipitates were immunoblotted for associated vimentin and NOD2, and direct lysates with HA-11 and vimentin antibodies. (D) SW480 cells were transfected with HA-NOD2 or HA-empty vector and cell extracts immunoprecipitated with vimentin-specific antibodies or non-specific antibody as control. Precipitates were immunoblotted for associated NOD2 and vimentin, and direct lysates with HA-11 and vimentin antibodies. Y2H, yeast-2-hybrid; WT, wild-type; IP, immunoprecipitate; mAb, monoclonal antibody; HA, hemagglutinin.
The LRR domain of NOD2 is the major determinant for binding to vimentin, and common CD-associated polymorphisms disrupt this interaction
The observation that the NOD2-R702W variant has reduced affinity for vimentin suggests that the LRR domain may be important for mediating this interaction. Using NOD2 mutants, NOD2 (1–693) lacking the C-terminal LRR domain and NOD2 (1–247) comprising only the two N-terminal CARDs (figure 2A), we demonstrated by immunoprecipitation that only full-length NOD2 can interact with vimentin (figure 2B), suggesting that the LRR domain contains the major determinant for binding to vimentin. To further determine whether the CD-associated polymorphisms affect the interaction with vimentin, we used cells expressing NOD2-R702W, NOD2-G908R and NOD2-L1007fs (figure 2A) to demonstrate that NOD2 and the NOD2-G908R variant can interact with vimentin to a similar degree (figure 2C). However, the NOD2-R702W and NOD2-L1007fs variant proteins seemingly lose their ability to interact with vimentin (figure 2C).

The leucine-rich repeat (LRR) domain contains the major determinant for binding to vimentin. (A) Schematic showing the structure of NOD2 and the location of Crohn's disease (CD)-associated polymorphisms. The NOD2 deletion mutants lacking the LRR domain or lacking both the LRR and NACHT (NAIP, CIITA, HET-E and TP1) domains generated by site-directed mutagenesis are also shown. (B) Haemagglutinin (HA)-NOD2 or the deletion mutants were transfected into HEK293 cells and cell lysates immunoprecipitated with vimentin-specific antibodies. Precipitates were immunoblotted for associated NOD2 proteins and vimentin, and direct lysates with HA-11 and vimentin antibodies. (C) HEK293 cells were transfected with HA-NOD2, HA-NOD2-R702W, HA-NOD2-G908R or HA-NOD2-L1007fs variants, and cell lysates immunoprecipitated with vimentin-specific antibodies. Precipitates were immunoblotted for associated NOD2 and vimentin, and direct lysates with HA-11 and vimentin antibodies.
Given that MDP also binds to the LRR domain of NOD2, we investigated whether stimulation with L18-MDP (a cell-permeable derivative of MDP) would affect the NOD2–vimentin interaction. Our immunoprecipitation results demonstrate that NOD2 interacts with vimentin to a similar degree in the absence or presence of MDP (online supplementary figure 1).
NOD2 interacts with vimentin at the plasma membrane, and common CD-associated variants are mislocalised to the cytosol
To determine the effect of the LRR-domain polymorphisms on the cellular localisation of NOD2, cells were transfected with HA-NOD2, R702W, G908R or L1007fs variants and immunostained for E-cadherin (basolateral plasma membrane) and HA-NOD2. We found that NOD2 and the G908R variant exhibited diffuse cytosolic staining but with distinct staining clearly observed at the plasma membrane (figure 3A, panels B, D and I, K, respectively, and quantified in figure 3B). In contrast, the R702W and L1007fs variants exhibited diffuse cytosolic staining with little or no distinct staining at the plasma membrane (figure 3A, panels F and M, respectively, and quantified in figure 3B). These results are in agreement with a previous study14 and confirm the importance of the LRR domain for the correct localisation of NOD2 to the plasma membrane.



Subcellular localisation of Crohn's disease (CD)-associated NOD2 variants. (A) SW480 cells were transfected with haemagglutinin (HA)-NOD2 (wild-type (WT)), HA-NOD2-R702W, HA-NOD2-G908R or HA-NOD2-L1007fs variants and immunostained with E-cadherin-specific antibodies (green) and exogenous NOD2 with HA-11 antibodies (red). Merged images demonstrate areas where NOD2 and E-cadherin colocalise (yellow) and are highlighted in HA-NOD2 and HA-NOD2-G908R cells (panels D and K). (B) Quantification of NOD2 membrane localisation in (A). Twenty cells from three separate fields of view were evaluated for membrane-localised NOD2. unpaired t test ***p<0.001. (C) HEK293 cells were transfected with HA-NOD2, HA-NOD2-R702W, HA-NOD2-G908R or HA-NOD2-L1007fs variants. After transfection, proteins were separated into specific subcellular fractionations. Each fraction was immunoblotted for NOD2 proteins, vimentin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), E-cadherin and histone H1. (D) Relative protein membrane fraction of HA-NOD2 (i), HA-NOD2-R702W (ii), HA-NOD2-G908R (iii) and HA-NOD2-L1007fs (iv) in relation to the cytosolic fraction using densitometry. Cyt, cytosol; Mem, membrane. (E) HEK293 cells were transfected with HA-NOD2 and immunostained with HA-11 antibodies (red) or vimentin-specific antibodies (green). Areas where NOD2 and vimentin colocalise are highlighted and appear as yellow.
To further confirm that the R702W and L1007fs variants result in altered cellular distribution of NOD2, cells were transfected with HA-NOD2 or the R702W, G908R or L1007fs variants, and cell lysates were purified into distinct subcellular compartments (figure 3C). The cytosolic to membrane ratio of NOD2 and G908R protein was 1:0.72 and 1:0.60, respectively (figure 3D, panels i and iii, respectively). In contrast, the R702W and L1007fs variant proteins both exhibited decreased membrane localisation, with cytosolic to membrane ratios of 1:0.38 and 1:0.31, respectively (figure 3D, panels ii and iv). Each compartment was immunoblotted with a specific marker: vimentin (cytoskeleton), GAPDH (cytosol), E-cadherin (membrane) and histone H1 (nucleus) as control. The observation that some L1007fs and R702W variant protein remains in the membrane fraction may be due to impurity of the fractionation process; however, our positive controls were clean, suggesting that NOD2 may localise to other sites in the cell such as the endoplasmic reticulum and the nuclear or mitochondrial membranes.
Two intriguing observations were that (1) the majority of NOD2 resided in the cytoskeletal fraction (figure 3C) and (2) vimentin could be detected in both the cytoskeletal and membrane fractions (figure 3C). These results further demonstrate that NOD2 can interact with cytoskeletal proteins such as vimentin, and suggest that NOD2 and vimentin may interact throughout the cell and at cell membranes. To test this hypothesis, cells were transfected with HA-NOD2 and immunostained for vimentin and HA-NOD2. Consistent with our fractionation results, vimentin was distributed throughout the cell and exhibited distinct staining around the nuclear and plasma membranes (figure 3E, panel A), with NOD2 exhibiting diffuse cytosolic staining but also with distinct staining at the plasma membrane (figure 3E, panel B). Merging of the two images shows clear areas of colocalisation between NOD2 and vimentin at the plasma membrane (figure 3E, panels C and D).
WFA disrupts the interaction between vimentin and NOD2 and relocalises NOD2 from the plasma membrane to the cytosol
The natural product WFA, a steroidal lactone from the plant Withania somnifera, binds to and inhibits vimentin.15 To confirm the effects of WFA in our experimental system, treatment of cells with 2 μM WFA was sufficient to induce vimentin aggregation and cleavage (figure 4A) with only minor effects on cell viability (figure 4B). Given that WFA causes vimentin to aggregate and retract from the cell periphery (figure 4C), we expected that WFA would disrupt the interaction between NOD2 and vimentin. Immunoprecipitation showed that WFA treatment significantly reduced the amount of NOD2 protein that interacts with vimentin (figure 4D). Furthermore, immunofluoresence microscopy demonstrated that WFA treatment resulted in the relocalisation of NOD2 from the plasma membrane to the cytosol (figure 4E and quantified in figure 4F). Taken together these results suggest that interaction with vimentin is important for the correct localisation of NOD2 to the plasma membrane.

Withaferin A (WFA) inhibits vimentin and relocalises NOD2 to the cytosol. (A) HEK293 cells were treated with WFA or dimethyl sulphoxide (DMSO) control as indicated for 2 h. Cell extracts were then immunoblotted for vimentin and actin. (B) The viability of cells treated with WFA or DMSO control for 2 h at the indicated concentrations was evaluated using trypan blue exclusion. (C) HEK293 cells were treated with WFA (2 μM) or DMSO control for 2 h. Cells were then immunostained with vimentin-specific antibodies and co-stained with 4′,6′-diamidino-2-phenylindole (DAPI) to detect nuclei. (D) HEK293 cells transfected with haemagglutinin (HA)-NOD2 were treated with WFA (2 μM) or DMSO control for 2 h, and cell lysates immunoprecipitated with vimentin-specific antibodies. Precipitates were immunoblotted for associated NOD2 and vimentin, and direct lysates with HA-11 and vimentin antibodies. (E) HEK293 cells were transfected with HA-NOD2. Cells were then treated with WFA (2 μM) or DMSO control for 2 h. After treatment, cells were immunostained with HA-11 antibodies and co-stained with DAPI to detect nuclei. (F) Quantification of NOD2 membrane localisation in (E). Twenty cells from three separate fields of view were evaluated for membrane-localised NOD2. unpaired t test ***p<0.001.
WFA inhibits NOD2-dependent NF-κB activation
NOD2 has been shown to form a complex with receptor interacting protein 2 and signal downstream to NF-κB from the plasma membrane.6 To assess whether the interaction with vimentin is important for this aspect of NOD2 activity, we performed luciferase reporter assays in SW480 colon epithelial cells, which express endogenous NOD2.6 Our results clearly demonstrate that treatment of cells with L18-MDP or TNFα stimulates the activity of a NF-κB-luciferase reporter plasmid (NF-κB-luc-WT), but not a control reporter plasmid with the NF-κB-binding sites deleted (NF-κB-luc-Mut) (figure 5A). Next, we performed the assay in the absence or presence of increasing concentrations of WFA. Again L18-MDP and TNFα treatments effectively stimulated NF-κB activity, and pretreatment with WFA inhibited activation of the reporter in a dose-dependent manner (figure 5B). WFA treatment was able to inhibit NF-κB activation by both L18-MDP and TNFα, a result that is consistent with a previous report that WFA can directly inhibit NF-κB DNA-binding activity and nuclear translocation.16 However, WFA was more effective at inhibiting L18-MDP- than TNFα-dependent NF-κB activation (figure 5B), suggesting some specificity of WFA on NOD2 activity. P65/RelA is a subunit of NF-κB that translocates from the cytosol to the nucleus on activation.17 To further confirm our findings, we monitored P65/RelA translocation in response to L18-MDP and TNFα treatments in the absence or presence of increasing concentrations of WFA. Pretreatment with WFA inhibited P65/RelA translocation by both L18-MDP and TNFα, but again was more effective at inhibiting L18-MDP-dependent NF-κB activation (online supplementary figure 2). These results suggest that interaction with vimentin and localisation to the plasma membrane are important for NOD2 ability to respond to MDP and signal downstream to NF-κB.

Withaferin A (WFA) inhibits NOD2-dependent nuclear factor-kappaB (NF-κB) signalling. (A) SW480 cells were transfected with NF-κB-luciferase wild-type (NF-κB-luc-WT) or mutant with NF-κB-binding sites deleted (NF-κB-luc-Mut) (200 ng) and the internal control Renilla (100 ng). After 16–18 h, cells were treated for 6 h with L18-muramyl dipeptide (MDP) (1 μg/ml) or for 2 h with tumour necrosis factor (TNF)α (50 ng/ml) and harvested for luciferase assays. The data are derived from triplicate readings. (B) SW480 cells were transfected with NF-κB-luciferase wild-type (NF-κB-luc-WT) or mutant with NF-κB binding sites deleted (NF-κB-luc-Mut) (200 ng) and the internal control Renilla (100 ng). After 16–18 h, cells were treated for 6 h with L18-MDP (1 μg/ml) or for 2 h with TNFα (50 ng/ml) in the presence of WFA (2 h) or dimethyl sulphoxide (DMSO) control (2 h) and harvested for luciferase assays. The data are derived from triplicate readings. unpaired t test *p<0.05; ns, not significant.
WFA inhibits NOD2-dependent autophagy
NOD2 has recently been shown to stimulate autophagy in response to MDP, and recruit the critical autophagy protein, ATG16L1, to the plasma membrane.18 ,19 To test whether vimentin regulates the autophagic activity of NOD2, we assessed the effect of WFA on NOD2-dependent autophagy stimulated by L18-MDP. HEK293 cells, which do not express endogenous NOD26 and were generated to stably express FLAG-EMPTY vector, FLAG-NOD2 or FLAG-NOD2-L1007fs (figure 6A,B), were transiently transfected with a plasmid to express the autophagy marker, LC3, fused to green fluorescent protein (GFP). LC3-I is converted into its membrane-associated lipidated form, LC3-II, a biochemical marker that correlates with the formation of autophagososmes and is commonly used to monitor autophagy levels.20 L18-MDP efficiently stimulated autophagy in HEK293-NOD2 cells, and this effect was inhibited by treatment with WFA (figure 6C,D). As a control, we assessed the effects of MDP in HEK293-L1007fs cells. As expected, L18-MDP had no effect on autophagy in these cells (figure 6E,F). As a further control, serum withdrawal, a potent stimulator of autophagy, resulted in a significant increase in LC3 punctae and in LC3-II in both HEK293-NOD2 and HEK293-L1007fs cells (figure 6C–F). These results are consistent with recent reports that plasma membrane localisation is important for NOD2 to activate autophagy and further establishes vimentin as an important regulator of NOD2 activity.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Withaferin A (WFA) inhibits NOD2-dependent autophagy. (A) HEK293 cells stably expressing FLAG-EMPTY, FLAG-NOD2 or FLAG-NOD2-L1007fs were generated. Cell extracts were immunoblotted with NOD2-specific antibodies. (B) HEK293 cells stably expressing FLAG-EMPTY, FLAG-NOD2 or FLAG-NOD2-L1007fs were immunostained with NOD2-specific antibodies. (C, E) HEK293 cells stably expressing FLAG-NOD2 (C) or FLAG-NOD2-L1007fs (E) were transfected to express green fluorescent protein (GFP)-LC3. Cells were pretreated for 30 min with WFA (2 μM) before treatment for 8 h with L18-muramyl dipeptide (MDP) (50 μg/ml). After treatment, the percentage of cells exhibiting more than five distinct LC3 punctate autophagosomes per cell was determined by fluorescence microscopy (i) and quantified in (ii). GFP-LC3-positive cells in five separate fields of view were counted for each sample. unpaired t test **p<0.01. (D, F) HEK293 cells stably expressing FLAG-NOD2 (D) or FLAG-L1007fs (F) were pretreated for 30 min with WFA (2 μM) before treatment for 8 h with L18-MDP (50 μg/ml). Cell extracts were immunoblotted for LC3 and actin. The ratio of LC3-II to LC3-I was measured by densitometry.
NOD2 and vimentin regulate the invasiveness and survival of AIEC
Recent evidence suggests that AIEC play a putative role in CD,4 and that NOD2 can limit the survival of intracellular bacteria.21 Therefore we compared the survival of the CD mucosa-associated AIEC strain, CUICD541-10,22 in HEK293-NOD2 and HEK293-L1007fs stable cells. As a control, we used the non-AIEC strain, CUH17-1. Our results show that NOD2 protects host cells by limiting the survival of CUICD541-10 and that this is defective in L1007fs-expressing cells (figure 7A). To assess the role of vimentin in the pathogenicity of the AIEC strain, CUICD541-10, we first compared its ability to invade HEK293 cells when vimentin was inhibited by preincubation of cells with an antibody that specifically binds to vimentin or with WFA. Our results clearly show that HEK293 cells express vimentin on the cell surface (online supplementary figure 3) and that blocking vimentin with specific antibody (compare figure 7B,C) or inhibiting vimentin with WFA (figure 7D) effectively limits host cell invasion by CUICD541-10 in a dose-dependent manner. To assess the importance of vimentin for NOD2 ability to limit bacterial survival, we infected HEK293-NOD2 stable cells with CUICD541-10 or CUH17-1 before treatment with increasing concentrations of WFA and further incubation. Our results show that WFA impairs the ability of NOD2 to limit bacterial survival in a dose-dependent manner (figure 7E).
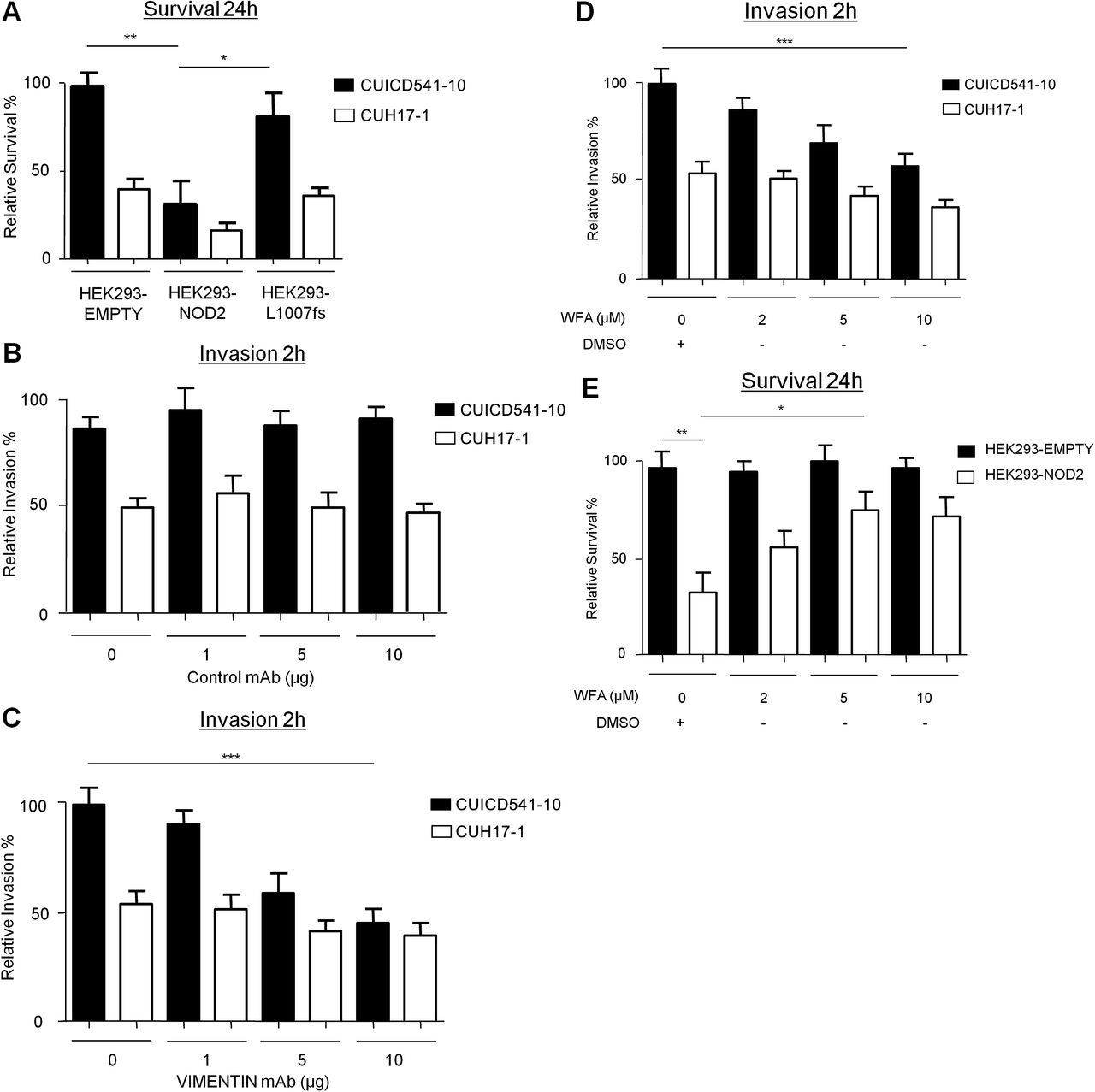
NOD2 and vimentin regulate the invasion and survival of a Crohn’s disease (CD)-associated Escherichia coli strains with an adherent and invasive phenotype (AIEC). (A) Bacterial survival was evaluated in HEK293 cells stably expressing FLAG-EMPTY, FLAG-NOD2 or FLAG-NOD2-L1007fs by gentamicin protection assay after 24 h using the E coli strain CUICD541-10 or CUH17-1. (B) Suppression of E coli strain CUICD541-10 or CUH17-1 invasion of HEK293 cells by preincubation of host cells with non-specific control antibodies, (C) antibodies that specifically block vimentin or (D) withaferin A (WFA) or dimethyl sulphoxide (DMSO) control was assessed by gentamicin protection assay after 2 h. (E) To assess the effect of WFA on E coli strain CUICD541-10 survival, bacteria were allowed to invade HEK293 cells stably expressing FLAG-EMPTY or FLAG-NOD2 before treatment with WFA or DMSO control. The effect of NOD2 and WFA on the survival of CUICD541-10 was assessed by gentamicin protection assay after 24 h. unpaired t test *p<0.05, **p<0.01, ***p<0.001.
The haplotype block containing Vim is a susceptibility locus for CD
A total of 965 SNPs in the haplotype block containing Vim and the two flanking blocks, spanning 163 kb, were tested. A total of 51 SNPs attained a p value of <0.001, with 47 of these in the block containing Vim. The strongest SNP (rs1049341) is positioned in the ninth exon (ENSE00001906870) of the Vim gene and attained a p value of 4.67×10−5. A plot of the SNPs in the region is presented in figure 8.

Results of a meta-analysis of seven Crohn's disease genome-wide association studies imputed with the 1000 genomes reference set. Scatterplot showing −log p values in the 163 kb region of Vim for 965 single-nucleotide polymorphisms. Vertical dotted lines represent the boundaries of the Vim gene, and solid lines the limits of each haplotype block. The −log p value corresponding to p<0.05 is represented by the horizontal dotted line.
Discussion
Since the discovery of NOD2 as a CD susceptibility gene,8 a substantial amount of both basic and clinical research has demonstrated several functions for NOD2, especially with regard to the innate immune response. Studies have now revealed important roles for NOD2 in viral recognition, regulation of the intestinal microbiome, and regulation of bacterial autophagy.19 ,23 ,24 Despite these advances in the understanding of NOD2 function, little is still known about how NOD2 exerts its effects at the molecular level or how polymorphisms present in the coding region of the LRR domain of NOD2 can lead to the development of CD and its associated complications.
Further to our previous work in a Y2H model,10 we now report that NOD2 and vimentin interact in mammalian cells and demonstrate the LRR domain of NOD2 as the major determinant for binding to vimentin. Importantly, we show that CD-associated polymorphisms in this domain disrupt this interaction. LRR domains are present in a large number of proteins, such as Toll-like receptors, and are important for the regulation of protein–protein interactions.25 The LRR domain of NOD2 is essential for sensing pathogens, with R702W, G908R and L1007fs variants defective in their ability to respond to bacterial lipopolysaccharide and peptidoglycan.26 Interestingly, our study demonstrates that the G908R variant retains the ability to bind vimentin, suggesting that distinct regions within the LRR domain mediate pathogen recognition and vimentin binding. This is consistent with our observation that treatment of cells with the peptidoglycan constituent, MDP, does not affect the NOD2–vimentin interaction.
LRR domains have also been shown to be important for targeting of proteins to cell membranes,27 and several studies have shown that NOD2 is localised to, and functions at, the plasma membrane,6 ,18 ,28 with the L1007fs protein reported to be mislocalised to the cytosol.6 In agreement with these studies, we observed localisation of wild-type NOD2 at the plasma membrane and L1007fs in the cytosol; in addition, we report the cytosolic mislocalisation of the R702W variant. Importantly, we show that NOD2 and vimentin colocalise at the plasma membrane and that mislocalisation of the L1007fs and R702W variants correlates with an inability to interact with vimentin. It is possible that the L1007fs and R702W polymorphisms cause a conformational change that makes the LRR domain inaccessible, thereby preventing the correct localisation of the protein. However, our observation that WFA treatment causes the relocalisation of NOD2 to the cytosol, together with our binding studies, strongly suggests that an inability to interact with vimentin contributes to the mislocalisation of L1007fs and R702W NOD2 variants. Significantly, we demonstrate that NOD2 activities that are dependent on its plasma membrane localisation, namely NF-κB activation, autophagy induction and bacterial handling, are disrupted by interference with the NOD2–vimentin interaction.
Manipulation of the host cell plasma membrane and associated proteins is an essential step for the uptake, survival and replication of pathogens.29 Several lines of research have demonstrated that vimentin is involved in this process. For example, cell-surface-expressed vimentin has been reported to be the binding target for several viruses, including porcine reproductive respiratory syndrome virus30 and Japanese encephalitis virus.31 Vimentin structure changes profoundly during bacterial infection, and upregulation of vimentin gene expression has been observed during pathogenic Helicobacter pylori infection.32 Most relevant to CD pathogenesis are the recent reports that vimentin is expressed on the surface of human brain endothelial cells, where it acts as a primary receptor for meningitis-associated E coli (MenPEC) strains expressing the virulence factor IbeA,11 with vimentin-mediated signalling required for IbeA+ E coli invasion of these cells.11 The IbeA virulence factor is not restricted to MenPEC, and is also present in a variety of other E coli pathovars, including 18.5% of AIEC strains and 17% of diarrheagenic E coli.22 ,33 Significantly, our results demonstrate that vimentin is important for invasion of host cells with a CD-associated AIEC and that inhibiting vimentin interferes with the ability of NOD2 to limit bacterial survival.
Our fractionation results unexpectedly revealed that the majority of NOD2 is bound to the cytoskeleton. NOD2 activity has been shown previously to depend on the integrity of the actin cytoskeleton,28 and drugs that disrupt actin have been shown to significantly increase the ratio of soluble to insoluble NOD2.28 Several bacterial pathogens manipulate the host cell cytoskeleton for their own intra- and inter-cellular motility,34 with the invasion of epithelial cells with AIEC markedly decreased by cytochalasin D and colichicine, indicating an important role for actin microfilaments and microtubules.22 An important role for the cytoskeleton in the regulation of autophagy is also emerging.35 Of particular significance are the findings that proper formation and distribution of autophagosomes depends on the integrity of IF networks36 and that autophagic vacuoles are tightly associated with vimentin.37 Our results showing that vimentin is important for NOD2 activity, together with recent studies linking NOD2 and the cytoskeleton with the regulation of autophagy, suggest that NOD2 has evolved to use the cytoskeletal network, in particular cell surface-expressed vimentin, as a means to engage with pathogens. NOD2 may then use the intracellular cytoskeleton further to directly transport internalised pathogens via the autophagy machinery to lysosomes for degradation.
Further studies are now required to address the vimentin–NOD2 interaction in patients with CD. Our imputed GWAS results further suggest that Vim is a susceptibility gene for CD, raising the possibility that vimentin is dysregulated in a pathway containing NOD2. Although our previous data showed that variations in Vim were not associated with CD susceptibility,10 this study only included four SNPs and was probably underpowered to detect an effect. Additional genetic studies are needed to confirm this association at a genome-wide significant level, including assessing epistasis between Vim and known susceptibility genes. In addition, in our previous micro-array study,10 we observed increased levels of vimentin expression in inflamed CD biopsy specimens compared with inflamed control biopsy samples, although it is important to note that our micro-array data do not discriminate vimentin expression between individual cell types. A focus of our current research is to further evaluate vimentin expression in mucosal biopsy tissue and peripheral blood and lamina propria mononuclear cell populations. As expected, our preliminary immunohistochemical staining of ileal sections using vimentin antibody (online supplementary figure 4) shows little or no expression of vimentin in intestinal epithelial cells; however, vimentin is highly expressed in cells throughout the lamina propria in both control and CD patient material. Significantly, using optical projection tomography, a new technique for three-dimensional imaging of small biological tissue samples, we observed vimentin expression at the base of small-intestinal crypts (online supplementary figure 5). Whether this involves the Paneth cells or epithelial stem cells needs to be explored further. However, given that NOD2 is highly expressed in this location,38 our finding is clearly directly pertinent. It is a well-recognised problem in the field that commercially available antibodies are unable to detect endogenous NOD2. Therefore we feel that (recognising these limitations) the use of an artificial overexpression system adequately answers our experimental questions. Once an antibody that can reliably detect endogenous NOD2 becomes available, further studies will be required to validate our key findings in tissue and primary cells from patients with CD.
In conclusion, this work, which describes a novel functional link between NOD2 and vimentin, together with the emerging role of vimentin in bacterial pathogenicity, identifies vimentin as a potential molecular target for therapeutic intervention for CD. We envisage that WFA could be targeted to a specific subgroup of patients with confirmed ileal CD with known AIEC colonisation either as a prophylactic or therapeutic modality. A major focus of our future research will be to assess whether modulation of vimentin represents a potential treatment for CD, and in this respect it is encouraging that WFA is already a well-recognised therapy, with three clinical trials currently registered on the WHO ICT Registry Platform (http://www.who.int/ictrp/en) looking at various health benefits in other conditions such as pulmonary tuberculosis.
Acknowledgments
We wish to thank Dr Charlie Lees and Dr Gwo-Tzer Ho for critical review of the manuscript. We also thank Dr Lesley Stark for technical assistance and reagents.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data Supplement 1 - Online Figure 1
- Data Supplement 2 - Online Figure 2
- Data Supplement 3 - Online Figure 3
- Data Supplement 4 - Online Figure 4
- Data Supplement 5 - Online Figure 5
- Data Supplement 6 - Online supplement 1
Footnotes
Access of researchers to data: All authors had open access to the raw dataset and statistical methods used during the preparation of the manuscript.
Funding CS is funded by a Medical Research Council project grant (No G0800759). PH is funded by a Medical Research Council project grant for PICTS (No G0800675).
Correction notice This article has been corrected since it was published Online First. The legend for figure 3A has been amended to read: (A) SW480 cells were transfected with haemagglutinin (HA)-NOD2 (wild-type (WT)), HA-NOD2-R702W, HA-NOD2-G908R or HA-NOD2-L1007fs variants and immunostained with E-cadherin-specific antibodies (green) and exogenous NOD2 with HA-11 antibodies (red).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement A summary of the complete dataset is available on request from Dr Craig Stevens at craig.stevens{at}ed.ac.uk.