Article Text

Abstract
Objective Hereditary pancreatitis is caused by mutations in human cationic trypsinogen (PRSS1) which lead to increased autoactivation by altering chymotrypsin C (CTRC)-dependent trypsinogen activation and degradation. Exceptions are some cysteine mutations which cause misfolding, intracellular retention and endoplasmic reticulum stress. Clinical relevance of many PRSS1 variants found in patients with sporadic chronic pancreatitis is unknown but often assumed by analogy with known disease-causing mutations. Functional comparison of PRSS1 variants found in sporadic and hereditary cases is needed to resolve this dilemma.
Design Here, we investigated the functional phenotype of 13 published PRSS1 variants with respect to autoactivation in the presence of CTRC and cellular secretion.
Results Only mutation p.D100H increased trypsinogen autoactivation, but this gain in function was offset by a marked reduction in secretion. Five mutants (p.P36R, p.G83E, p.I88N, p.V123M, p.S124F) showed decreased autoactivation due to increased degradation by CTRC. Five mutants exhibited strongly (p.D100H, p.C139F) or moderately (p.K92N, p.S124F, p.G208A) reduced secretion, whereas mutant p.K170E showed slightly increased secretion. Mutant p.I88N was also secreted to higher levels but was rapidly degraded by CTRC. Finally, three mutants (p.Q98K, p.T137M, p.S181G) had no phenotypic alterations relative to wild-type trypsinogen.
Conclusions Rare PRSS1 variants found in sporadic chronic pancreatitis do not stimulate autoactivation but may cause increased degradation, impaired secretion or no functional change. Variants with reduced secretion are likely pathogenic due to mutation-induced misfolding and consequent endoplasmic reticulum stress.
- PANCREAS
- PANCREATIC DISEASE
- PANCREATIC DISORDERS
- PANCREATIC ENZYMES
- PANCREATITIS
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Hereditary pancreatitis is caused by mutations in human cationic trypsinogen (PRSS1) that increase autoactivation.
-
Some pathogenic PRSS1 variants misfold, suffer intracellular retention and cause endoplasmic reticulum stress.
-
Clinical relevance of rare PRSS1 variants found in subjects with sporadic chronic pancreatitis is uncertain but frequently assumed.
What are the new findings?
-
Rare PRSS1 variants found in sporadic chronic pancreatitis do not increase trypsinogen activation.
-
Rather, they cause increased degradation, loss of secretion or no functional change.
-
Reduced secretion is indicative of misfolding that may increase pancreatitis risk through endoplasmic reticulum stress.
How might it impact on clinical practice in the foreseeable future?
-
Functional classification of PRSS1 variants found in patients with chronic pancreatitis is required for determination of clinical relevance.
Introduction
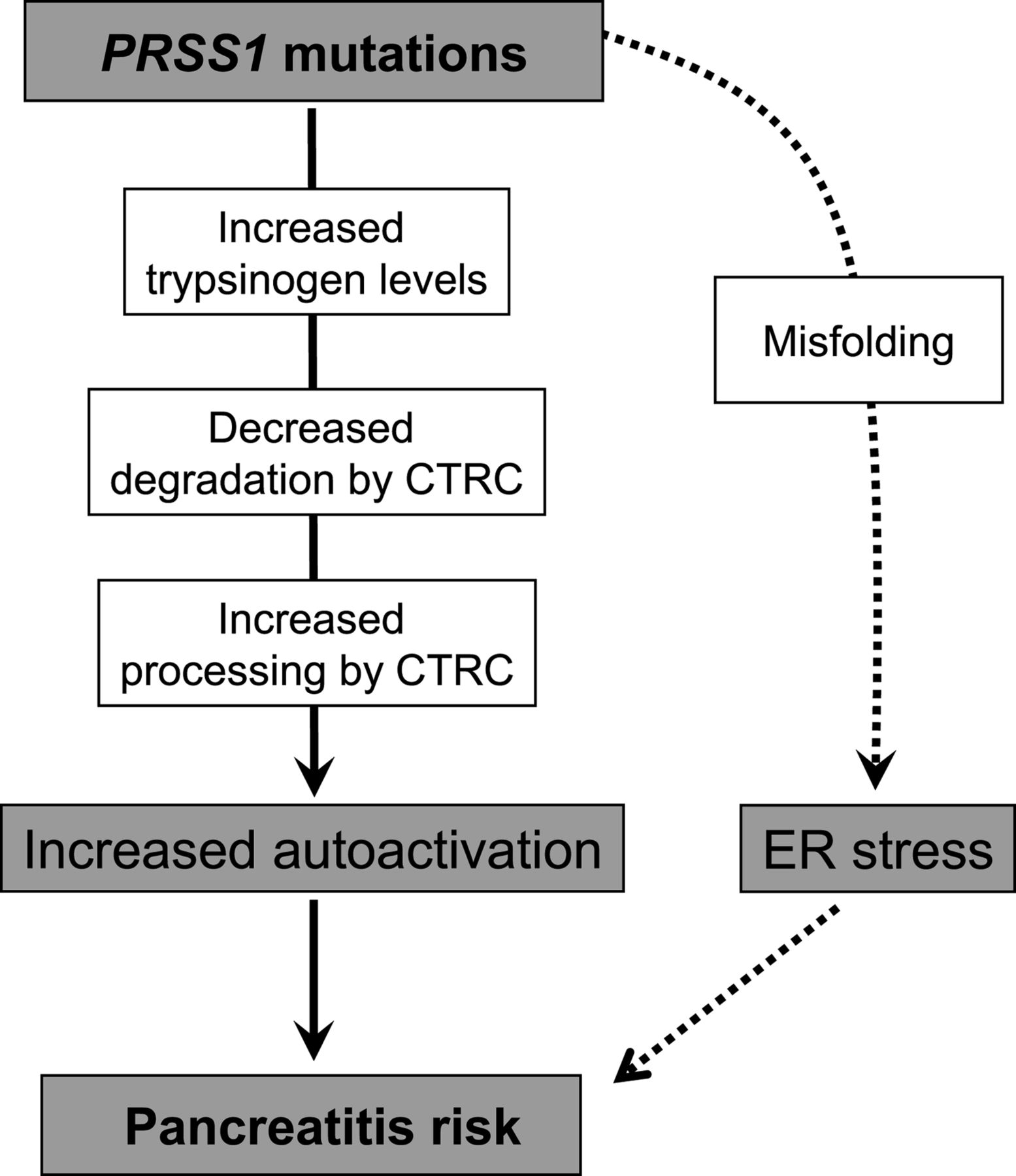
Hereditary pancreatitis is an autosomal dominant disorder caused by mutations in the serine protease 1 (PRSS1) gene that codes for cationic trypsinogen.1 Causative mutations are p.R122H (∼65%), p.N29I (∼25%) and less frequently p.A16V, p.D22G, p.K23R, p.K23_I24insIDK, p.N29T, p.V39A, p.R116C and p.R122C.1–5 To date, 21 additional rare missense PRSS1 variants have been reported, the majority of which were found in patients with sporadic chronic pancreatitis with no family history5 (http://www.pancreasgenetics.org). The mechanism of action of hereditary pancreatitis-associated mutations involves increased autoactivation of mutant trypsinogens resulting in elevated intrapancreatic trypsin activity levels6 (figure 1). Recent studies uncovered that PRSS1 mutations alter the regulation of activation and degradation of cationic trypsinogen by chymotrypsin C (CTRC). The digestive enzyme CTRC stimulates trypsinogen activation by processing the activation peptide to a shorter form, which is easier cleaved by trypsin.7 Somewhat paradoxically, CTRC also promotes degradation of trypsinogen by cleaving the calcium binding loop.6 ,8 This cleavage in combination with a trypsin-mediated autolytic cleavage results in inactivation of trypsinogen during autoactivation and lower trypsin levels attained. Pancreatitis-associated mutations render trypsinogen resistant to CTRC-dependent degradation and/or increase N-terminal processing by CTRC and thereby elevate trypsin levels generated through autoactivation6 (figure 1).

Pathological pathways associated with PRSS1 mutations in hereditary and sporadic chronic pancreatitis. Mutations in PRSS1 can increase autoactivation of cationic trypsinogen by different mechanisms: increased trypsinogen expression or secretion; inhibition of chymotrypsin C (CTRC)-dependent trypsinogen degradation, stimulation of N-terminal processing of the trypsinogen activation peptide by CTRC; and direct stimulation of autoactivation. Alternatively, PRSS1 mutations can cause misfolding and endoplasmic reticulum (ER) stress. See text for further details.
The unifying pathological mechanism described above does not seem to apply to some mutations that alter the number of cysteine residues in cationic trypsinogen. Hereditary pancreatitis-associated mutation p.R116C was shown to induce protein misfolding with intracellular retention and degradation, which may represent an alternative disease-causing mechanism unrelated to trypsinogen activation and trypsin activity.9 Mutation p.C139S, which was reported in sporadic cases of chronic pancreatitis, exhibits similar properties.9 Mutation-dependent misfolding can elicit endoplasmic reticulum (ER) stress, which might be responsible for increased pancreatitis risk, although the mechanism remains unclear (figure 1).
In the present study, we surveyed the functional properties of 13 rare missense PRSS1 variants found in patients with sporadic chronic pancreatitis. Our primary objective was to test whether these variants also exhibit increased activation in the presence of CTRC as previously seen with disease-causing mutants in hereditary pancreatitis. A second objective of the study was to assess cellular secretion of the mutants to determine whether mutation-induced changes in folding and secretion may be a more common phenotype of PRSS1 variants than previously appreciated.
Experimental procedures
Nomenclature
Amino acid residues in human cationic trypsinogen (serine protease 1, PRSS1) are numbered starting with the initiator methionine of the primary translation product in accordance with the recommendations of the Human Genome Variation Society.
Plasmid construction and mutagenesis
The pTrapT7 intein-PRSS1, pcDNA3.1(-) PRSS1 and pcDNA3.1(-) CTRC 10His expression plasmids were constructed previously.7 ,8 ,10 Missense mutations were introduced by overlap extension PCR mutagenesis, cloned into the expression plasmids and verified by DNA sequencing.
Expression and purification of trypsinogen
Wild-type and mutant trypsinogens were expressed in the aminopeptidase P deficient LG-3 Escherichia coli strain as fusions with a self-splicing mini-intein, as described in.10 ,11 This expression system was developed to produce recombinant trypsinogen with uniform, authentic N-termini. Isolation of cytoplasmic inclusion bodies, in vitro refolding and purification with ecotin affinity chromatography were carried out according to published protocols.10 ,11 Mutant p.C139F could not be purified by this method, as it misfolded during in vitro refolding. Concentrations of trypsinogen preparations were calculated from their ultraviolet absorbance at 280 nm using the extinction coefficient 37 525 M−1 cm−1.
Cell culture and transfection
Human embryonic kidney 293T (HEK 293T) cells were cultured and transfected as described previously.12 Transfections were performed using 1 µg expression plasmid and 2.5 µl Lipofectamine 2000 (Invitrogen, Carlsbad, California, USA) in 2 ml Dulbecco's Modified Eagle Medium. After overnight incubation, cells were washed and the transfection medium was replaced with 2 ml OPTI-MEM I Reduced Serum Medium (Invitrogen) containing 1 mM benzamidine (final concentration) to inhibit autoactivation of secreted trypsinogen. Conditioned media were harvested 24 h after addition of OPTI-MEM.
Expression and purification of human CTRC
Large-scale expression of human CTRC in HEK 293T cells and purification from the conditioned medium using nickel-affinity chromatography were performed as reported previously.6 CTRC was activated with human cationic trypsin and active CTRC concentrations were determined by active site titration with ecotin, as described.13
Trypsinogen autoactivation in the presence of CTRC
Trypsinogen at 1 µM concentration was incubated with 5 nM human CTRC and 10 nM cationic trypsin in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 0.05% Tween-20 (final concentrations) at 37°C. At given times, 2 µl aliquots were withdrawn and mixed with 48 µl assay buffer containing 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 0.05% Tween-20. Trypsin activity was measured by adding 150 µl 200 µM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate and following the release of the yellow p-nitroanilin at 405 nm in a SpectraMax plus384 microplate reader (Molecular Devices, Sunnyvale, California, USA) for 1 min. Reaction rates were calculated from fits to the initial linear portions of the curves. The trypsin substrate was dissolved in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 0.05% Tween-20.
Measurement of trypsin activity in conditioned media
Aliquots (50 µl) of conditioned media were supplemented with 5 µl 1 M Tris-HCl (pH 8.0) and 1 µl 0.5 M CaCl2 and trypsinogens were activated by adding 1 μl 1.4 μg/ml human enteropeptidase (R&D Systems, Minneapolis, Minnesota, USA). After incubation for 1 h at 37°C, a 50 µl aliqout was removed and mixed with 150 µl 200 µM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate. Activity was determined as described above.
Sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) and western blotting
Conditioned media (200 µl) was precipitated with 10% trichloroacetic acid (final concentration), resuspended in 20 μl Laemmli sample buffer containing 100 mM dithiothreitol, heat-denatured at 95°C for 5 min and run on 15% SDS-polyacrylamide gels. The gels were stained with Coomassie Blue R-250. For western blotting, 5 µl conditioned media were directly mixed with sample buffer and electrophoresed as described above. Proteins were transferred onto an Immobilon-P membrane (Millipore Corporation, Bedford, Massachusetts, USA) at 300 mA for 1.5 h. The membrane was blocked with 5% milk powder dissolved in phosphate-buffered saline supplemented with 0.1% Tween-20 (final concentration), at 4°C overnight. Trypsinogen was detected with a sheep polyclonal antibody (R&D Systems, #AF3848) used at a dilution of 1:5000 followed by horse-radish peroxidase (HRP)-conjugated donkey polyclonal antisheep IgG (R&D Systems, #HAF016) used at 1:2000 dilution. Incubations with primary and secondary antibodies were performed at room temperature for 1 h each. HRP was detected using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Quantitation of bands was carried out with the Image Lab 3.0 (Bio-Rad) software.
Results
PRSS1 variants found in subjects with sporadic chronic pancreatitis
We studied here 13 missense PRSS1 variants found in patients with idiopathic chronic pancreatitis or recurrent acute pancreatitis with no family history (table 1). The variants were detected in the heterozygous state, with the exception of p.G208A, which was also identified in a homozygous individual. As indicated, the variants were found only in one to three cases each; therefore, the genetic information was insufficient to determine whether these variants are pathogenic or neutral. Unaffected carrier parent was reported in two cases, for variant p.C139F and p.S181G. Note that variants p.T137M, p.K170E and p.G208A were from subjects of Asian origin, whereas all other variants were from subjects of European origin.
Rare PRSS1 variants in pancreatitis
To determine whether these 13 variants are prevalent in the general population, we consulted the NHLBI Exome Sequencing Project Exome Variant Server (http://evs.gs.washington.edu) which lists aggregate exome-sequencing data for approximately 4300 European-American and 2200 African-American individuals. We found PRSS1 variants p.V123M and p.T137M in one European-American subject each and variant p.S181G in one African-American subject. However, interpretation of these findings is difficult, because the database also lists the disease-causing mutation p.R122C found in two European-American individuals, suggesting that the studied cohort may have contained pancreatitis patients. Therefore, we sequenced exon-3 of the PRSS1 gene in 1000 German subjects without any pancreatic disease. Exon-3 was selected because most of the studied variants (nine of 13) were in this region. With the exception of a novel c.367G>T (p.V123L) variant found in one subject, no other alterations were identified. This observation is in agreement with published sequencing data showing the absence of these 13 PRSS1 variants in 200 French,14 82 German,15 420 Chinese16 ,17 and 28 Korean18 control subjects. We conclude that the 13 PRSS1 variants presumed to cause chronic pancreatitis (table 1) are not generally found in a healthy population.
Autoactivation and catalytic activity of PRSS1 variants
First, trypsinogen variants were characterised for their ability to autoactivate in the absence of CTRC. When measured at pH 8.0, in 1 mM Ca2+, only one mutant, p.D100H, exhibited increased autoactivation, which was about 2.5-fold faster than wild type (see online supplementary figure S1A). Five mutants (p.Q98K, p.T137M, p.K170E, p.S181G and p.G208A) autoactivated as well as wild-type cationic trypsinogen (see online supplementary figure S1B). Mutants p.K92N and p.S124F autoactivated at a decreased rate but reached essentially the same final trypsin levels as wild-type trypsinogen (see online supplementary figure S1C). In contrast, four mutants exhibited decreased rates of autoactivation (p.P36R, p.G83E, p.I88N and p.V123M) with reduced final trypsin levels, suggesting that these mutants may become partially degraded during autoactivation (see online supplementary figure S1D).
Catalytic activity of the activated trypsin variants was measured with the synthetic peptide substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide. As shown in online supplementary table S1, enzyme kinetic parameters of trypsin variants were comparable with those of wild-type cationic trypsin. This finding is consistent with previous observations that natural PRSS1 variants almost never affect catalytic function of trypsin.
Note that mutant p.C139F could not be purified in sufficient quantities due to misfolding; therefore, it was not tested in the autoactivation and catalytic activity assays.
Autoactivation of PRSS1 variants in the presence of CTRC
To determine the effect of CTRC on the trypsinogen variants, we measured autoactivation in the presence of 5 nM human CTRC. This low CTRC concentration slightly stimulates the rate of autoactivation but causes a reduction in final active trypsin levels due to trypsinogen degradation during activation.6 In the experiments presented, wild-type cationic trypsinogen reached about 50% activity in the presence of CTRC, relative to trypsin activity observed in the absence of CTRC (not shown, also see figure 1A in6). Under these conditions, both increases and decreases in autoactivation are readily detectable.
Only one of the 12 variants tested, p.D100H, exhibited increased activation in the presence of CTRC (figure 2A). A similar phenotype was observed with this mutant when autoactivation was performed in the absence of CTRC (see above), indicating that the mutation increases autoactivation independent of CTRC. Consistent with this interpretation, degradation of p.D100H mutant trypsinogen and trypsin by CTRC was unchanged (not shown). Six mutants (p.K92N, p.Q98K, p.T137M, p.K170E, p.S181G and p.G208A) autoactivated in the presence of CTRC in a manner that was comparable with wild type (figure 2B). Surprisingly, however, five mutants (p.P36R, p.G83E, p.I88N, p.V123M and p.S124F) reached markedly reduced trypsin activity levels during autoactivation, suggesting increased susceptibility to CTRC-dependent degradation (figure 2C). This notion was confirmed by direct degradation experiments which demonstrated that these five mutants were degraded by CTRC at increased rates (see online supplementary figure S2).

Autoactivation of cationic trypsinogen variants in the presence of chymotrypsin C (CTRC). Trypsinogen was incubated at 1 μM concentration with 5 nM CTRC and 10 nM initial trypsin in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 and 0.05% Tween-20 (final concentrations) at 37°C. At the indicated times, 2 μl aliquots were removed and trypsin activity was measured as described in Experimental procedures section. Trypsin activity was expressed as per cent of the maximal wild-type activity in the absence of CTRC. (A) Mutant with increased autoactivation. (B) Mutants with unchanged autoactivation. (C) Mutants with decreased autoactivation.
Secretion of PRSS1 variants from HEK 293T cells
To identify trypsinogen variants defective in folding, we evaluated their secretion from transfected cells to the conditioned medium using SDS-PAGE with Coomassie Blue staining, western blot analysis and trypsin activity measurements after activation with enteropeptidase. The use of HEK 293T cells for cellular secretion studies is a compromise, as efficient transfection of pancreatic acinar cells is not feasible. Despite its constitutive secretion, this cell line remains a good model to study ER related disturbances such as misfolding, retention and degradation of secretory proteins.
Six of 13 mutants tested (p.P36R, p.G83E, p.Q98K, p.V123M, p.T137M and p.S181G) showed trypsinogen secretion close to wild-type levels (∼70%–85%) (figure 3A–C, black bars). Severe secretion defects (∼20% of wild type) were observed with mutants p.D100H and p.C139F. Moderate reduction in secretion (∼40%–50% of wild type) was noted with three mutants (p.K92N, p.S124F and p.G208A). Two mutants (p.I88N and p.K170E) showed increased secretion levels, 140% and 130% of wild type, respectively. The higher secretion of mutant p.I88N was due to the appearance of a second trypsinogen band which represents an aberrantly glycosylated trypsinogen form (figure 3A,B). This band was also observed in a much fainter form with wild-type trypsinogen and all other variants and may be an artefact of the heterologous cell line used. Although increased secretion of variant p.I88N may represent a gain of function, the rapid degradation of this variant by CTRC (see above) would cancel out this effect and result in a loss-of-function phenotype. On the other hand, it seems probable that the slightly more efficient secretion of mutant p.K170E may confer an increased risk for pancreatitis.

{kind=link}
{kind=link}
{kind=link}
Secretion of cationic trypsinogen variants from HEK 293T cells. Cells were transiently transfected and conditioned media were collected after 24 h. Trypsinogen protein levels were qualitatively analysed on Coomassie Blue stained SDS-polyacrylamide gels (A) and then quantitatively determined by western blots (B) and densitometry (C, black bars). Trypsin activity was measured after activation with enteropeptidase (C, grey bars). See Experimental procedures section for details. Representative gels and blots of four independent experiments are shown. Average of four experiments with SD was plotted. Trypsinogen protein and trypsin activity levels were expressed as per cent of wild-type values.
For the majority of the variants, trypsin activity levels in the conditioned medium correlated well with the protein levels secreted (figure 3C, grey bars). Four variants (p.P36R, p.G83E, p.K92N and p.C139F) had considerably lower enzyme activity relative to their protein levels. Variant p.C139F may be misfolded and catalytically defective, which explains its lower activity, whereas mutants p.P36R and p.G83E suffer some degradation during activation, as seen in the autoactivation experiments in online supplementary figure S1D. The lower activity of mutant p.K92N is unexplained and may be related to increased sensitivity to some inhibitory component in the cell culture medium, because when this mutant was purified either from E coli or from the conditioned medium of transfected HEK 293T cells, wild-type trypsin activity levels were reached after activation with enteropeptidase.
Discussion
In this study, we investigated the functional properties of 13 rare PRSS1 variants detected in patients with sporadic chronic pancreatitis. The common biochemical phenotype of PRSS1 mutations associated with hereditary pancreatitis is the generation of greatly increased trypsin levels during autoactivation in the presence of CTRC.6 This phenomenon is due to increased sensitivity of trypsinogen mutants to CTRC-mediated stimulation of autoactivation and/or resistance to CTRC-dependent trypsinogen degradation (figure 1). Unexpectedly, only one of the investigated 13 mutants exhibited a similar phenotype when autoactivation was tested in the presence of CTRC. Mutant p.D100H autoactivated at a faster rate and reached higher trypsin levels than wild-type trypsinogen; however, this was independent of CTRC as the same phenotype was observed in its absence. More importantly, further studies, discussed below, demonstrated that mutant p.D100H was poorly secreted, which would completely negate any increased propensity for autoactivation and result in diminished trypsin levels upon activation.
Five mutants were degraded by CTRC at an increased rate resulting in lower trypsin levels during autoactivation, relative to wild-type trypsinogen. This biochemical phenotype is inconsistent with the trypsin-dependent model of hereditary pancreatitis and suggests that these variants are not pathogenic (table 2). Previous studies found that PRSS1 mutations p.R116C and p.C139S resulted in reduced trypsinogen secretion.9 These mutations alter the number of Cys residues and thus may interfere with correct disulfide formation during folding, which results in intracellular retention and degradation. A similar secretion defect, likely caused by mutation-induced misfolding, was observed in the present study for variants p.D100H and p.C139F and, to a smaller extent, variants p.K92N, p.S124F and p.G208A, suggesting that these variants are pathogenic (table 2). Trypsinogen misfolding underlying the reduced secretion may elicit ER stress and thereby increase the risk for pancreatitis, as previously shown for mutants p.R116C and p.C139S9 (figure 1). Secretion defect and ER stress was also observed for a subset of CTRC mutants associated with chronic pancreatitis.12 ,19 Increased secretion of mutant trypsinogens may result in higher trypsinogen levels in the pancreatic juice with consequently increased risk for autoactivation (figure 1). Copy number mutations in trypsinogen genes may exert their pathogenic effect via this mechanism.20 ,21 Conversely, decreased trypsinogen expression due to the c.1-408C>T polymorphism in the 5′ region is associated with decreased pancreatitis risk.22 In this study, we observed slightly increased secretion with two mutants, p.I88N and p.K170E. While the rapid degradation of mutant p.I88N by CTRC would counteract the effect of higher trypsinogen concentrations, the phenotype of mutant p.K170E may indicate a true gain of function with potentially elevated risk of pancreatitis (table 2). Finally, three trypsinogen variants (p.Q98K, p.T137M and p.S181G) proved functionally neutral in this study, indicating that PRSS1 variants of no clinical significance may be incidental findings when subjects with chronic pancreatitis are screened for underlying genetic defects (table 2).
Clinical relevance of PRSS1 variants classified on the basis of functional phenotype
In summary, functional analysis of 13 PRSS1 variants found in patients with chronic pancreatitis demonstrated that these rare PRSS1 variants do not phenocopy the disease-causing hereditary pancreatitis-associated PRSS1 mutations. Instead, increased degradation or reduced secretion was observed mostly. Variants with decreased secretion may increase pancreatitis risk through an alternative pathological pathway related to mutation-induced misfolding rather than trypsin activity.
Acknowledgments
These studies were supported by NIH grants R01DK058088, R01DK082412, R01DK082412-S2 and R01DK095753 (to MS-T). AS was also supported by a fellowship from the Rosztoczy Foundation in Boston and AS and PH were supported by the TÁMOP-4.2.2/B-10/1-2010-0012 grant in Szeged. The authors thank Vinciane Rebours (Beaujon Hospital, Clichy, France) and Jian-Min Chen (Université de Bretagne Occidentale, Brest, France) for providing unpublished information.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors The study was designed by AS, PH and MS-T. The experiments were performed by AS, SB and HW. The manuscript was written by AS, SB, PH and MS-T.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.