Article Text

Abstract
Objective To investigate the potential tumour suppressor functions of glutathione peroxidase 7 (GPX7) and examine the interplay between epigenetic and genetic events in regulating its expression in oesophageal adenocarcinomas (OAC).
Design In vitro and in vivo cell models were developed to investigate the biological and molecular functions of GPX7 in OAC.
Results Reconstitution of GPX7 in OAC cell lines, OE33 and FLO-1, significantly suppressed growth as shown by the growth curve, colony formation and EdU proliferation assays. Meanwhile, GPX7-expressing cells displayed significant impairment in G1/S progression and an increase in cell senescence. Concordant with the above functions, Western blot analysis displayed higher levels of p73, p27, p21 and p16 with a decrease in phosphorylated retinoblastoma protein (RB), indicating its increased tumour suppressor activities. On the contrary, knockdown of GPX7 in HET1A cells (an immortalised normal oesophageal cell line) rendered the cells growth advantage as indicated with a higher EdU rate, lower levels of p73, p27, p21 and p16 and an increase in phosphorylated RB. We confirmed the tumour suppressor function in vivo using GPX7-expressing OE33 cells in a mouse xenograft model. Pyrosequencing of the GPX7 promoter region (−162 to +138) demonstrated location-specific hypermethylation between +13 and +64 in OAC (69%, 54/78). This was significantly associated with the downregulation of GPX7 (p<0.01). Neither mutations in the coding exons of GPX7 nor DNA copy number losses were frequently present in the OAC examined (<5%).
Conclusions Our data suggest that GPX7 possesses tumour suppressor functions in OAC and is silenced by location-specific promoter DNA methylation.
- CANCER
- CELL CYCLE
- GASTROINTESTINAL CANCER
- OESOPHAGEAL CANCER
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
-
Glutathione peroxidase 7 (GPX7) is a recently identified member of the GPX family.
-
Earlier studies have shown that GPX7 is often downregulated in oesophageal adenocarcinomas (OAC).
-
GPX7 protects oesophageal epithelial cells against acidic bile salt-induced oxidative DNA damage, double-strand breaks and cell death through reduction of intracellular reactive oxygen species (ROS) level.
-
Loss of GPX7 promotes accumulation of intracellular ROS and oxidative DNA damage.
What are the new findings?
-
This study demonstrates that GPX7 can regulate the cellular growth pattern and the levels of p27, p21 and p16 and phospho-RB proteins—key cell cycle regulators.
-
In addition, GPX7 regulates the levels of p73, an important family member of the tumour suppressor p53.
-
Reconstitution of GPX7 expression in OAC cells suppresses their tumour growth capacity in a xenograft mouse model.
-
GPX7 dysfunction in OAC is predominantly regulated by promoter DNA hypermethylation, in particular the CpG sites from +13 to +64, rather than the genetic mutation and copy number loss.
How might it impact on clinical practice in the foreseeable future?
-
This study provides direct evidence for the role of GPX7 in oesophageal tumorigenesis, highlighting previously unknown tumour suppressor functions of GPX7.
-
Taken together with our previous studies, GPX7 is a dual function protein that has potent antioxidant and tumour suppressor functions that might have an important role in protecting the genomic integrity of oesophageal cells. Therefore, it is possible that patients with loss of GPX7 may be at higher risk of progression towards oesophageal adenocarcinoma.
Introduction
The incidence of oesophageal adenocarcinomas (OAC) has increased by 4–10% a year among men since 1976, more rapidly than for any other type of cancers.1 Barrett's oesophagus (BO), a precancerous condition in which the original oesophageal squamous epithelia are replaced by columnar epithelia owing to chronic gastro-oesophageal reflux disease, is the main risk factor for development of OAC.2–4 Patients with BO can progress to low-grade dysplasia, high-grade dysplasia and OAC at 30–60 times that of the general population.4–6 The natural and molecular history of the progression of BO to OAC remains poorly characterised.
Glutathione peroxidase (GPX) is a major antioxidant enzyme family that catalyses the reduction of hydrogen peroxide (H2O2), organic hydroperoxide and lipid peroxides by reduced glutathione.7 ,8 The GPX family comprises eight members: GPX1–GPX8. GPX7 was first identified from cDNAs amplified from mRNA of Brca1-null mouse embryonic fibroblasts.9 Recently, GPX7 was shown to have limited glutathione peroxidase activity, although it can neutralise H2O2 in vitro without the existence of glutathione.10 Expression of GPX7 can protect normal oesophageal epithelia from acidic bile salt-induced oxidative stress, oxidative DNA damage and double strand breaks.10 Frequent dysfunction of GPX7 in OAC and its precancerous Barrett's dysplasia11 suggests that impairment of the antioxidant capacity may contribute to the development of OAC. However, the full spectrum of the molecular functions of GPX7 has not been established. In this study, we examined the possible tumour suppressor function of GPX7 in vitro and in vivo and determined the possible epigenetic and genetic mechanisms that might regulate its expression in OAC.
Materials and methods
Cell lines
Three immortalised cell lines originating from normal oesophageal squamous epithelia: HEEC (ScienCell Research Laboratories, Carlsbad, California, USA), EPC2 (kindly provided by Dr Hiroshi Nakagawa) and HET1A (purchased from American Type Culture Collection (ATCC), Manassas, Virginia, USA); four immortalised cell lines originating from BO: BAR-T, BAR-T10 (kindly provided by Dr Rhonda Souza), CP-A and CP-B (purchased from ATCC) and lastly, five oesophageal adenocarcinoma cell lines were used in this study: OE19 (purchased from Sigma-Aldrich, St Louis, Missouri, USA), OE33, FLO-1, SKGT4 and JH-ESO-Ad1 (a gift from Drs David Beer and Jim Eshleman). FLO-1, OE33 and HET1A were used in the functional experiments while other cell lines were used in gene expression, DNA methylation, DNA copy number and mutation analysis. The HEK293 AD cell line (Cell Biolabs Inc, San Diego, California, USA) was used for propagating adenoviral particles. All cell lines were grown at 37°C in 5% carbon dioxide following the recommended procedures.
Tissue samples
All de-identified tissue samples were obtained from the archives of pathology at Vanderbilt University (Nashville, Tennessee, USA) and from the National Cancer Institute Cooperative Human Tissue Network. The use of specimens from the tissue repository was approved by the Vanderbilt institutional review board. For DNA and mRNA analysis, 148 frozen tissue samples (78 OAC, six BO, 36 normal oesophagus and 28 normal stomach samples) were collected. All adenocarcinomas were classified according to the recent guidelines of the International Union Against Cancer TNM classification system. All OAC originated from the lower oesophagus or gastro-oesophageal junction corresponding to AEG type 1 as previously described.12 The patients’ ages ranged from 34 to 82 years (median 63). The adenocarcinomas ranged from well differentiated to poorly differentiated, stages I–IV, with a mix of intestinal and diffuse-type tumours.
Cloning and construction of GPX7 expression plasmids
A full length of GPX7 coding sequence with Flag-tag was amplified from normal cDNA by PCR and was cloned into the pcDNA 3.1 and pACCMV.pLpA plasmids.10 The GPX7-expressing adenoviral system was then constructed as previously described.13 Two oesophageal adenocarcinoma cell lines (FLO-1 and OE33) in which GPX7 expression was significantly downregulated, were transfected with control or GPX7-pcDNA plasmids using Lipofectamine 2000 transfection reagent (Life Technologies, Grand Island, New York, USA), or were infected with a multiplicity of infection (MOI) of 25 per cell of adenoviral GPX7 particles (Ad-GPX7) and adenoviral empty particles (Ad-CTRL) in the culture medium. Forty-eight hours after infection, the cells were harvested and validated for the expression of GPX7 using quantitative RT-PCR and Western blotting.
Knockdown of GPX7 expression by lentiviral shRNA
To knockdown GPX7 expression in non-malignant cells, the oesophageal squamous cell line HET1A was transfected with the same MOI of scrambled short hairpin RNA controls (Sc shRNA) and GPX7-validated specific shRNA (GPX7 shRNA) lentiviral particles as previous described.13
Determination of cell growth curve
FLO-1 and OE33 cells were infected with control or GPX7-expressing adenoviral particles or transfected with control or GPX7-expressing pcDNA plasmid (cells were under selection of 600 mg/ml G418 for 2 weeks). HET1A cells were transfected with Sc shRNA and GPX7 shRNA. Cell numbers were counted at 24 h intervals using the Bio-Rad TC10 Automated Cell Counter (Bio-Rad, Hercules, California, USA) with trypan blue exclusion assay. All experiments were performed in triplicate.
Colony formation assay
FLO-1 cells were infected with 25 MOI control or GPX7-expressing adenoviral particles. Forty-eight hours after infection, cells were split and seeded at a density of 500 cells/well in the six-well plates. Cells were cultured at 37°C for another 2 weeks. Cells were then stained with 0.5% crystal violet solution. The images of the plates were analysed using ImageJ software (NIH).
Soft agar colony formation assay
To check whether GPX7 is also involved in anchorage-independent growth, soft agar colony formation assay14 was performed. In brief, FLO-1 and OE33 cells were infected with 25 MOI control or GPX7-expressing adenoviral particles. Forty-eight hours after infection, cells were split and seeded at a density of 1.0×105 cells/well in a 0.35% noble agar (Sigma) mixed with culture medium which was topped on a bottom agar (0.5% noble agar with medium) in the six-well plates. Cells were cultured at 37°C for another 2 weeks. The images of the plates were captured under a microscope and were analysed using ImageJ software (NIH).
Cell proliferation assay
To measure cell proliferation, the Click-iT EdU Assay (Life Technologies) was performed following the manufacturer's recommendation, as previously described.15 EdU (5-ethynyl-2′-deoxyuridine) is a nucleoside analogue of thymidine and is incorporated into DNA during active DNA synthesis. EdU-positive cells (20 random fields at ×40, >400 cells) were counted using ImageJ software.
Flow cytometry analysis of cell cycle progression after synchronisation
To examine whether GPX7 expression impairs cell cycle progression, OE33 cells were infected with Ad-CTRL and Ad-GPX7 viral particles. Cells were blocked with 2 mM thymidine for 16 h, released from the block (cultured in full medium without thymidine) for 9 h and then blocked with 2 mM thymidine again for 16 h.16 Cells were released from the block and returned to full culture medium, harvested and fixed in 100% ethanol. They were then incubated with 40 µg/ml propidium iodide and 100 µg/ml RNase A at 37°C for 30 min and immediately subjected to fluorescent activated cell sorting analysis.
Detection of cell senescence
β-galactosidase activity was determined using a senescence β-galactosidase staining kit (Cell Signaling, Danvers, Massachusetts, USA) following the manufacturer's protocol. In brief, OE33 and FLO-1 cells were infected with control and GPX7-expressing adenoviral particles, cells were then split into six-well plates and cultured in serum-reduced medium (1% fetal bovine serum). At 24, 48 and 72 h after infection, cells were fixed and incubated with β-galactosidase staining solution (pH 6.0) at 37°C overnight in a dry incubator without CO2. The next day, the plates were checked and 10 ×200 fields were photographed. The β-galactosidase staining intensity was determined using ImageJ software (NIH).
Western blotting analysis
Western blot analysis was performed using standard protocols.15 The protein concentration was determined by a Bio-Rad protein assay using a FLUO Star OPTIMA microplate reader (BMG). The primary antibodies were anti-GPX7 antibody (rabbit, 1:1000, ProteinTech Group, Chicago, Illinois, USA), anti-p73 antibody (rabbit, 1:500, Bethyl, Montgomery, Texas, USA), anti-p21 antibody (mouse, 1:1000, Cell Signaling), anti-p27 antibody (rabbit, 1:1000, Cell Signaling), anti-p16 antibody (rabbit, 1:1000, Cell Signaling), anti-RB antibody (mouse, 1:1000, Cell Signaling), anti-phospho-RB, ser780 (rabbit, 1:1000, Cell Signaling), anti-phospho-RB, ser807 (rabbit, 1:1000, Cell Signaling) and anti-actin antibody (rabbit, 1:1000, Cell Signaling). Horseradish peroxidase-conjugated anti-mouse (1:10 000 dilution) and anti-rabbit (1:10 000 dilution) secondary antibodies were purchased from Cell Signaling Technology.
Xenografting in nude mice
To confirm GPX7 function in vivo, OE33 cells stably expressing GPX7 or empty pcDNA vector were injected subcutaneously into 6-week-old Nu/Nu nude mice (Charles River, Wilmington, Massachusetts, USA); 2×106 cells per injection site (10 sites per group). Tumour masses were monitored and measured twice a week and the tumour volume was calculated using the formula: Tvol=½ (L×W2) where Tvol is tumour volume, L is tumour length and W is tumour width. All mice were killed when the control group had tumours reaching a volume of 1000 mm3. The tumours were weighed and photographed. All animal experiments were performed in accordance with institutional guidelines and were approved by the animal care review board at the University of Vanderbilt.
Analysis of mRNA expression and DNA copy numbers of GPX7
Total RNA and DNA were isolated using the RNeasy and DNeasy mini kit (Qiagen, Valencia, California, USA). Single-stranded complementary DNA was subsequently synthesised from RNA using the iScript cDNA synthesis kit (Bio-Rad). The sequence of GPX7 and HPRT cDNA primers was described previously.11 The forward and reverse primers for GPX7 genomic DNA were 5′-GTGGAGGCAGGTAGAAGCTG-3′ and 5′-CAGGATCCCAGAAAAGTCCA-3′, respectively. The primers were obtained from Integrated DNA Technologies (Coralville, Iowa, USA). Quantitative real-time PCR (qPCR) was performed using an iCycler (Bio-Rad) with the threshold cycle number determined by the use of iCycler software V.3.0. The mRNA expression results were normalised to the average value of HPRT1, whereas the DNA copy number results were normalised to the average value of both β-actin and glyceraldehyde-3-phosphate dehydrogenase.17 Loss of DNA copy number was considered at a relative cut-off ratio of ≤0.5, whereas copy number gain was considered at cut-off ratio of ≥1.3. The fold expression was calculated as previously reported.11
DNA bisulfite treatment and pyrosequencing analysis
The DNA was modified by bisulfite using an EZ DNA methylation gold kit (Zymo Research, Orange, California, USA), according to the manufacturer's protocol. Five pyrosequencing assays were designed using PSQ assay design software (Qiagen) to cover a long region of GPX7 promoter from −162 to +138, relative to transcription start site (TSS) as shown in figure 8A and online supplementary table S1. The PCR conditions and subsequent pyrosequencing procedure were done according to established protocols.11 Based on control normal samples and internal quality controls provided in the software analysis, we used 10% as a cut-off value for identification of DNA hypermethylation.11
Statistical analysis
Data are expressed as the mean±SE of the mean for parametric data. An unpaired Student t test was performed for two independent samples. One-way analysis of variance with post-test comparison was used to analyse the independent samples of three or more. Spearman's rank correlation analysis was used to analyse the correlation between GPX7 methylation level and gene expression. All statistical analyses were done using GraphPad Prism4 software. For all analyses, p≤0.05 is considered statistically significant.
Results
Reconstitution of GPX7 suppressed growth of OAC cells in vitro
Using stable (pcDNA-GPX7) and transient (adenoviral) reconstitution models of GPX7 in OAC cell lines (FLO-1 and OE33), we detected a significant reduction in growth rates in GPX7-expressing cells (figure 1A–D). Concordant with these results, the reconstitution of GPX7 resulted in a significant suppression of colony formation in FLO-1 cells (figure 1E,F). For further validation, we performed a soft agar assay using both OE33 and FLO-1 cells and found that reconstitution of GPX7 led to significantly fewer and smaller colonies (p<0.01) (figure 2A,B). These data indicate that GPX7 can suppress both anchorage-dependent and anchorage-independent growth of OAC cells.

Reconstitution of glutathione peroxidase 7 (GPX7) expression suppresses oesophageal adenocarcinoma (OAC) cell growth in vitro. (A–D) Growth curves were plotted based on cell counting using trypan blue assay after reconstitution of expression of GPX7 in FLO-1 (A, B) and OE33 (C, D) OAC cell lines. Both pcDNA stable (A, C) and transient adenoviral (Ad) (B, D) reconstitution of GPX7 demonstrate significant reduction in cell growth, as compared with controls (p<0.01 at 96 and 120 h points). Western blots are included to demonstrate the levels of GPX7. (E, F) Colony formation assay in 500 OAC cells (FLO-1) using Ad (E) or pcDNA stable (pcDNA) (F) reconstitution of GPX7 demonstrate significant reduction in the number and size of colonies (p<0.01). Quantification is shown in the right panels. Ad-CTRL, adenoviral empty particles.
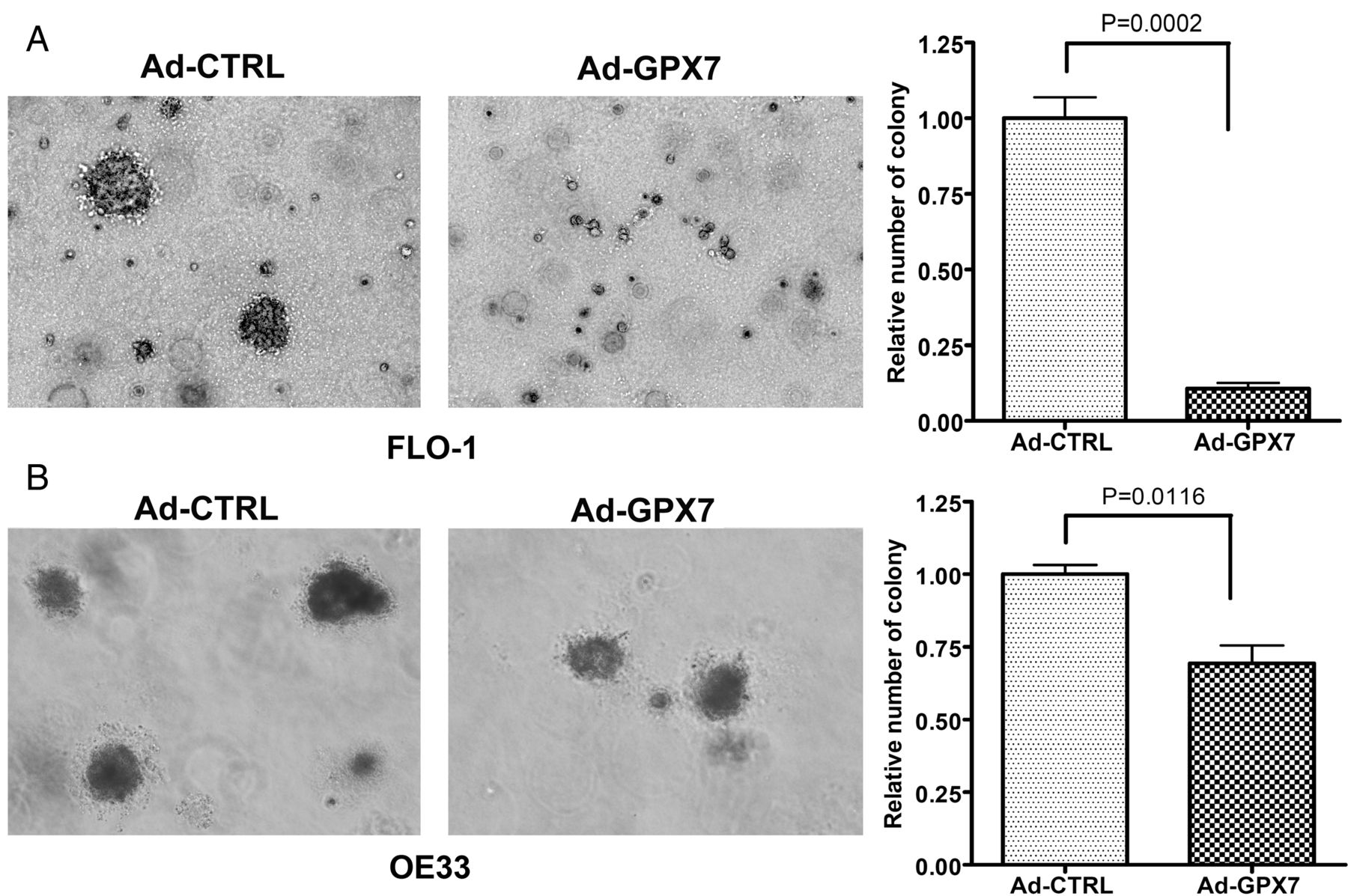
Glutathione peroxidase 7 (GPX7) suppresses anchorage-independent growth. (A, B) Soft agar colony formation assay in FLO-1 cells (A) and OE33 cells (B). In both (A, B), cells with GPX7-expressing adenoviral particles (Ad-GPX7) formed significantly smaller and fewer colonies than control (Ad-CTRL) (p<0.01). Quantification of the data is shown in the right panels.
Reconstitution of GPX7 in OAC cells suppressed tumour cell proliferation and impaired cell cycle progression
Because the aforementioned data suggested a possible effect on cell proliferation, we performed an EdU assay and found that OAC cells expressing GPX7 (Ad-GPX7) have a significantly lower positive rate for EdU incorporation than Ad-CTRL (p=0.038) (OE33 cells are shown in figure 3A, FLO-1 cells are shown in online supplementary figure S1). These findings prompted us to further examine the cell cycle. We synchronised OE33 cells at the G1/S point by thymidine double block,16 followed by release and analysis of cell cycle progression using fluorescent activated cell sorting. As shown in figure 3B, about half of the control cells (Ad-CTRL) had proceeded to the middle S phase after 4 h while only 32.4% of GPX7-expressing cells (Ad-GPX7) proceeded to the early S phase, suggesting that G1/S progression was delayed in GPX7-expressing cells.

Glutathione peroxidase 7 (GPX7) expression inhibits tumour cell proliferation and delays the G1/S progression. (A) EdU fluorescence staining (green) demonstrates a significant decrease of nuclear incorporation of EdU in OE33 cells following the expression of GPX7-expressing adenoviral particles (Ad-GPX7), in comparison with control cells (Ad-CTRL) (p=0.038). The right panel displays the quantitative data. (B) Flow cytometry analysis of cell cycle progression of OE33 cells after double thymidine block and release for 4 h; results show that about half of the control cells (Ad-CTRL) had proceeded to middle S phase after 4 h while only 32.4% of Ad-GPX7 proceeded to the early S phase. Representative flow cytometry histograms and quantification graphs are shown.
GPX7 promotes cell senescence in OAC cells in vitro
One of the key features of tumour suppressor genes is cellular senescence. Our analysis demonstrated that GPX7-expressing OAC cells (OE33 and FLO-1) had a significant increase in β-galactosidase staining (p≤0.01), as compared with control cells (figure 4), suggesting that GPX7 can promote cell senescence in OAC cells.

Glutathione peroxidase 7 (GPX7) promotes cell senescence in oesophageal adenocarcinoma cells in vitro. (A, B) Representative images of β-galactosidase staining of GPX7-expressing (Ad-GPX7) and control (Ad-CTRL) OE33 (A) and FLO1 (B) cells at 72 h following serum deprivation (1% fetal bovine serum) (p≤0.01). The right panels display quantitative results of the β-galactosidase staining intensity at 24, 48 and 72 h time points of serum deprivation (1% fetal bovine serum).
GPX7 expression deregulates cell cycle and senescence components of cell signalling
As we observed growth suppression, impairment in G1/S progression and cell senescence in GPX7-expressing OAC cells, we next examined cellular signalling components involved in these processes. Western blot analysis of FLO-1 and OE33 OAC cells that are stably expressing GPX7 (figure 5A) or after infection with GPX7-expressing adenoviral particles (figure 5B) showed an increase in the levels of p73, p27, p21 and p16. Of note, the p16 protein was not detectable in FLO-1 cells, suggesting that it may be deleted or methylated in this cell line, a common finding in oesophageal cancers. The decrease in the phosphorylation levels of the RB protein at serine 807 and serine 780 are indicative of increased RB activity in GPX7-expressing cells. These data provide a plausible explanation consistent with the biological phenotype that we observed after reconstitution of GPX7.

Glutathione peroxidase 7 (GPX7) regulates the protein levels of tumour suppressor genes. (A, B) Western blot analysis is shown for FLO-1 and OE33 oesophageal adenocarcinoma cells that are stably expressing GPX7 (5A) or after infection with GPX7-expressing adenoviral particles (Ad-GPX7) (B). An increase in the levels of p73, p27, p21 and p16 and a decrease in phosphorylated RB (p-RB) were detected (FLO-1 cell line is silent for p16).
Knockdown of GPX7 expression in HET1A cells promoted cellular proliferation
To confirm the tumour suppressor functions of GPX7, we knocked down GPX7 in non-malignant HET1A cells, which expresses a higher level of GPX7. As shown in figure 6, knockdown of GPX7 resulted in a significant increase in EdU rate (A) and cell growth rate (B). Western blotting analysis (C) confirmed the decrease in the levels of p73, p21, p27 and p16 and increase in the levels of phosphorylated retinoblastoma (RB) in GPX7 knockdown cells (GPX7 shRNA) as compared with that in control cells.

Knockdown of glutathione peroxidase 7 (GPX7) expression renders cell growth advantage. HET1A (an immortalised normal oesophageal cell line) cells were stably transfected with GPX7-specific shRNA (GPX7 shRNA) and control scramble shRNA (Sc shRNA). (A) EdU cell proliferation assay displays that knockdown of GPX7 expression (GPX7 shRNA) led to a significant increase in EdU positive rate, indicating a higher cellular proliferation rate. The left panel shows the representative EdU and DAPI images and the right panel shows the quantification using ImageJ software. (B) Growth curve shows a faster growth rate of GPX7-knockdown cells than control cells (p<0.05 at 72, 96 and 120 h time points). (C) Western blotting analysis demonstrates a reduction in the levels of p73, p27, p21 and p16 proteins and an increase of phospho-RB after GPX7 knockdown in HET1A cells.
Overexpression of GPX7 inhibited OAC tumour growth in vivo
To confirm the above results in vitro, we applied an in vivo xenografting mouse model. As shown in figure 7, the control OE33 cells generated large tumour masses, whereas the GPX7-expressing OE33 cells either failed to generate tumour mass or generated significantly smaller tumours (p=0.004). Of note, we performed a similar in vivo experiment using GPX3 reconstitution and failed to detect a similar tumour suppression effect (see online supplementary figure S2), suggesting that the tumour suppressor function is unique for GPX7.

Reconstitution of glutathione peroxidase 7 (GPX7) expression in oesophageal adenocarcinoma cells suppresses tumour growth in vivo. (A) OE33 cells stably expressing GPX7 or empty pcDNA vector (control) were subcutaneously xenografted into Nu/Nu nude mice. Representative images are shown. (B) Tumour volume was monitored and plotted as shown (*p<0.05; **p<0.01). (C) A comparison of the weight of resected tumours is shown. GPX7-expressing tumours were significantly smaller than controls (p=0.004).
Location-specific promoter DNA methylation is responsible for GPX7 downregulation in OAC
Gene mutations and allele alterations such as DNA copy number loss are involved in the downregulation of some tumour suppressor genes. However, we did not discover any missense mutation in the CDS region in cell lines originating from normal squamous epithelia (HEEC), BO (BAR-T), oesophageal adenocarcinoma (FLO-1, OE33, SKGT4 and JH-ESO-Ad1) and 10 OAC samples and their matched normal oesophagus (data available). Similarly, we did not detect DNA copy number losses of GPX7 in cell lines and OAC samples as compared with normal samples (p>0.05, see online supplementary figure S3 and table S1). These data suggested that genetic events do not play a major role in regulating GPX7 expression in OAC.
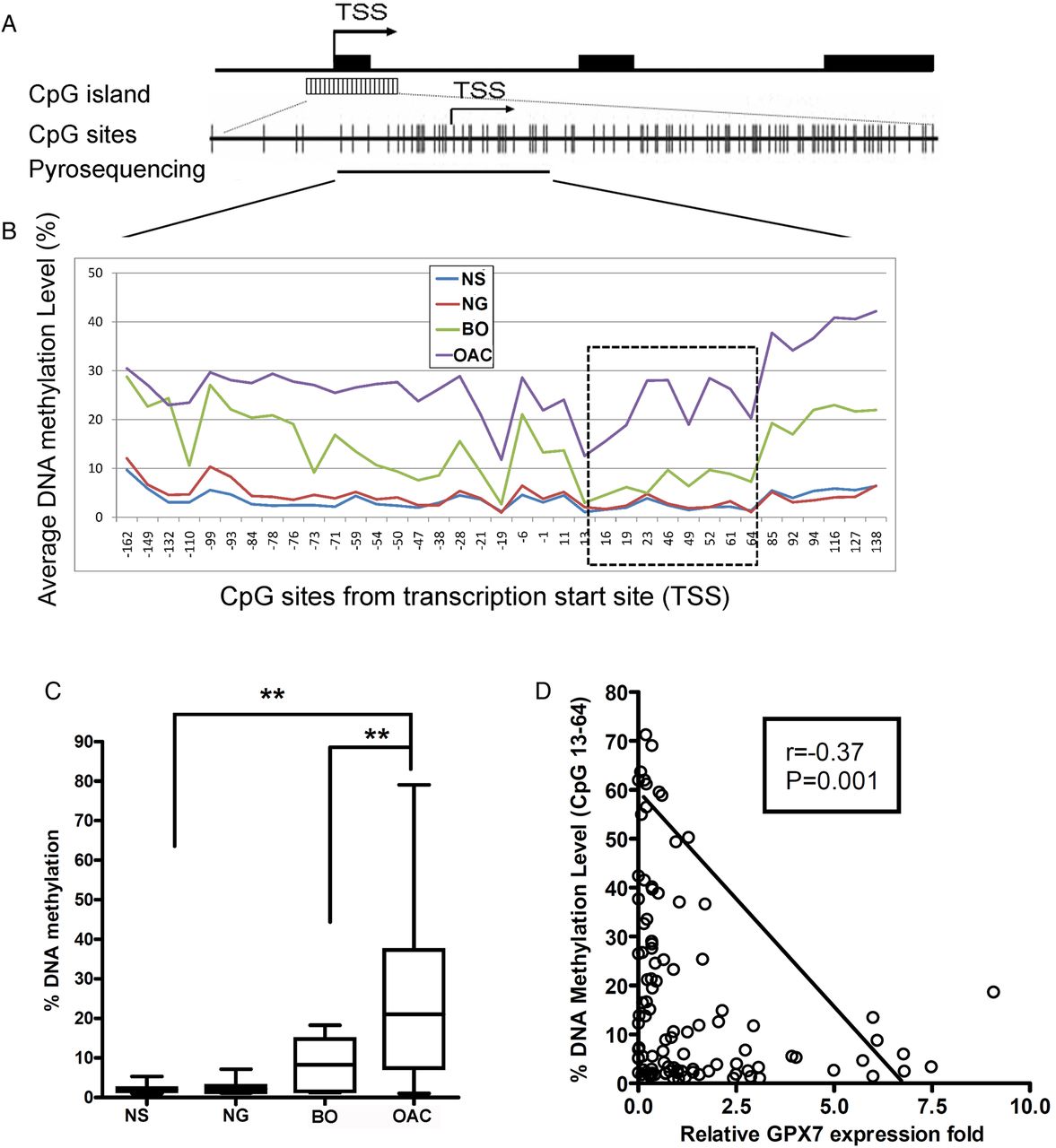
We have previously shown that methylation of GPX7 can regulate its expression.11 However, this earlier analysis was limited to a small region of the GPX7 promoter and did not take into consideration the recent observations that methylation may have location-specific effects.18–20 Our analysis of the GPX7 promoter spanned the region from −162 to +138, relative to TSS (figure 8A and see online supplementary table S1) and demonstrated low methylation levels (<10%) in normal squamous epithelia of the oesophagus (NS) and normal glandular epithelia of the stomach (NG, figure 8B). The BO demonstrated hypermethylation (≥10%) in all examined CpG sites except for sites from +13 to +64, suggesting that hypermethylation of GPX7 could start as early as BO. On the other hand, we found that hypermethylation of the CpG sites from +13 to +64 was unique to OAC samples (p<0.01) (figure 8B,C). Of note, methylation levels of this region inversely correlated with GPX7 expression (r=−0.37, p=0.001) (figure 8D), suggesting that location-specific methylation (+13 to +64) plays a critical role in regulating GPX7 expression. Interestingly, in cell lines CP-B, OE19, OE33 and some primary OAC (table 1), we observed that GPX7 promoter methylation levels (≥20%) can override the observed DNA copy number gains (≥1.3 fold change) in these samples, leading to downregulation of GPX7. Taken together, the results suggest that location-specific promoter hypermethylation is the major event in regulating GPX7 expression.
GPX7 DNA copy number, methylation level and mRNA expression in oesophageal cell lines and primary tumours
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Location-specific DNA methylation of glutathione peroxidase 7 (GPX7) promoter regulates GPX7 expression. (A) A schematic chart shows GPX7 genomic structure. GPX7 has three exons as shown in black boxes. A CpG island with dense CpG sites is present around the transcription start site (TSS); each vertical bar represents one CpG site. Pyrosequencing assays that covered CpG sites from −162 to +138, relative to TSS were designed (see online supplementary table S1). (B) Summary of pyrosequencing results in normal oesophageal squamous epithelia (NS), normal gastric mucosa (NG), Barrett's oesophagus (BO) and oesophageal adenocarcinoma (OAC) (additional details in table 1). Hypermethylation of CpG sites from +13 to +64 (dash-line square) was only seen in OAC. (C) Comparison of the average DNA methylation levels (CpG sites from +13 to +64) in NS, NG, BO and OAC samples. (D) Spearman correlation analysis demonstrated inverse correlation between methylation levels (+13 to +64) and GPX7 mRNA expression fold (r=−0.37, p=0.001).
Discussion
GPX7 is a relatively new member of the GPX family, which has not been fully characterised. We have recently shown that GPX7 can neutralise H2O2 and protect oesophageal epithelia from acidic bile salt-induced oxidative DNA damage by reducing intracellular reactive oxygen species levels.10 Because of the frequent silencing of GPX7 in OAC, we decided to examine its possible tumour suppressor functions. In this study, using transient and stable reconstitution of GPX7 in both in vitro and in vivo models of OAC, we have shown that GPX7 can suppress cellular proliferation and anchorage-dependent and -independent growth capacity, promote cellular senescence and mediate a delay in G1/S progression. On the contrary, knockdown of GPX7 expression promoted cell growth and cellular proliferation in non-malignant oesophageal cells. Although GPX7 promoter hypermethylation can occur at different promoter regions, our detailed analyses have demonstrated location-specific DNA promoter hypermethylation as the major event in regulating GPX7 dysfunction in OAC. Taken together, the results suggest that GPX7 possesses tumour suppressor functions that are lost through location-specific promoter methylation in OAC.
The regulation of the cell cycle is a complex cellular process where the balance between several cyclin-dependent kinases and their inhibitors dictates the ultimate outcome for the cell. Deregulation of these factors leads to loss of control of cellular proliferation, a fundamental feature of tumour development.21 ,22 The loss of protein expression of p16, p21 and p27 has been shown to be one of the steps taking place during progression from BO to OAC, leading to significant impairment of cell cycle control.23 ,24 The loss of p16 is considered to be one of the early molecular features of the development of OAC.25 ,26 p16, encoded by cyclin-dependent kinase inhibitor 2A (CDKN2A), is a classic tumour suppressor gene that plays a critical role in negatively regulating the cell cycle.27 ,28 Deletions, mutations and promoter methylation of p16 are among the most commonly described mechanisms for silencing its expression and functions in human cancer.27
In addition to the loss of p16 expression, inactivation of the cyclin-dependent kinase inhibitor, p27, has been shown in several tumour types, including OAC.29 p27 plays an important role in establishing a threshold for G1 cyclin/CDK accumulation before activation of CDK2 kinase and entry into the mitotic cycle.29 ,30 This finding is consistent with our results showing a delay in the G1/S progression following reconstitution of GPX7 where p27 becomes upregulated.
One of the key downstream targets of p53 is p21, a protein known to play critical roles in promoting cell cycle arrest.31 ,32 p21 is a potent cyclin-dependent kinase inhibitor which regulates the cell cycle progression at G1 by binding to, and inhibiting the activity of, CDK2 or CDK1 complexes. We are showing here that GPX7 can modulate the levels of p21 in OAC cell lines in which the p53 gene is mutant.33 This finding indicates that GPX7 can regulate the levels of p21 in a p53-independent mechanism and adds to the tumour suppressor capabilities of GPX7. Of note, we have shown here that one of the p53 family members, p73, is upregulated upon expression of GPX7 in tumour cells. Like p53, p73 has similar tumour suppressor functions and shares many p53 target genes such as p21.34 ,35 Therefore, the upregulation of p21 in GPX7-expressing cells was probably regulated through p73. In support of this, our knockdown results clearly showed that loss of GPX7 expression led to the loss of expression of p73 and p21, as well as p27 and p16, accompanied by increased phospho-RB levels that increased the cells’ growth advantage. We have previously shown that dysfunction of GPX7 is common and probably an early event occurring through BO to dysplasia stages.13 Taken together, these results suggest that dysfunction of GPX7 may play a crucial role in Barrett's tumorigenesis through deregulation of the key cell cycle regulators listed above.
Cellular senescence is one of mechanisms by which tumour suppressor genes exert their functions.36 ,37 We observed a significant increase in β-galactosidase staining intensity in GPX7-expressing cells as compared with control cells in both OE33 and FLO-1 cells (figure 5). It has been shown that p21 and p16, and RB genes, are key regulators of the cell cycle and senescence processes.36–38 Our western blotting results, which showed an increase in the levels of p21 and p16 and a reduction in the levels of phospho-RB at the sites of serine 807 and serine 708 in GPX7-expressing cells as compared with controls, support the cellular senescence phenotype observed. These data demonstrate that GPX7 may perform its tumour suppressor function, at least partially, by promoting tumour cell senescence.
The above results demonstrated the activation of key tumour suppressor genes upon reconstitution of GPX7 in OAC, but it was essential to determine whether the biological functions can be replicated in vivo and if they are specific to GPX7 or extend to other GPX family members. Previous studies have shown that GPX3 and GPX7 are the only GPX family members that are frequently deregulated in OAC.11 ,39 Contrary to our findings showing that reconstitution of GPX7 suppressed OAC tumour growth, reconstitution of GPX3, a GPX family member that is also methylated in OAC,11 ,39 failed to suppress tumour growth in vivo. These results suggest that the tumour growth suppressor functions are GPX7-specific. However, it is possible that other members of the GPX family may have functions in other cancers similar to those of GPX7.
Aberrant DNA methylation is one of the most common epigenetic alterations that regulate tumour suppressor genes in cancer cells.40–42 Recent evidence has suggested that not all CpG sites are functionally equal.18 ,20 Our earlier report analysing a limited region of the GPX7 promoter (−38 to +11 from TSS) suggested that methylation might regulate GPX7,11 but the extent of promoter methylation and functional relevance remained unclear. Furthermore, the possibility that genetic events might also lead to dysfunction of GPX7 were not explored. Several reports have shown that transcriptional silencing may be a result of the interaction of genetic and epigenetic events,17 ,43–45 but we did not detect mutations or copy number losses of GPX7 in OAC. Our comprehensive analysis of the GPX7 promoter CpG island in this study has highlighted the region from +13 to +64 as a specific region associated with OAC and was significantly inversely correlated with GPX7 expression, confirming that it is indeed a functional region of the GPX7 promoter. This finding is in line with recent publications showing that location-specific methylation of promoter regions has a crucial role in regulating gene transcription in cancer.18–20 This is further supported by our observation of downregulation of GPX7 in OAC samples with relative copy number gains, where promoter hypermethylation was present (table 1).
In conclusion, we have shown that GPX7 has tumour suppressor functions through regulating key cell cycle signalling molecules. Location-specific promoter methylation mediates GPX7 silencing and the loss of its potential tumour suppressor capacity, which might be a key step in unleashing the oncogenic features driving OAC.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors DFP: biological assays, methylation analysis and manuscript writing. TLH: PCR experiments. MS: assisted in animal studies. AB: experimental design, troubleshooting, review of the written draft. AZ: troubleshooting, interpretation of results. WE-R: study design, interpretation of results, troubleshooting, manuscript writing.
-
Funding This study was supported by grants from the National Institute of Health; R01CA106176, Department of Veterans Affairs, Vanderbilt SPORE in Gastrointestinal Cancer (P50 CA95103), Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute, Department of Veterans Affairs, or Vanderbilt University.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The authors will share reagents listed in this publication in accordance with Vanderbilt University and National Institute of Health guidelines of sharing reagents.