Article Text

Abstract
Background: Small bowel cancer (SBC) is one of the tumours associated with Lynch syndrome (LS). To advise on screening for this tumour it is paramount to be informed about the lifetime risk. The aim of this study was to calculate the lifetime risk of SBC in LS and to identify possible risk factors.
Methods: Clinical and pathological data were collected on 1496 proven or putative carriers of a mismatch repair gene mutation from 189 families. Kaplan-Meier survival analysis was used to calculate the lifetime risk and to assess potential risk factors.
Results: 28 (1.9%) of the 1496 (putative) mutation carriers were identified with SBC. The median age at diagnosis was 52 years (range 23–69 years). The lifetime risk of developing SBC was 4.2%. There was no difference in risk between males and females (log rank: p = 0.2470), or between MLH1 and MSH2 mutation carriers (log rank: p = 0.2754). SBC was not observed in MSH6 mutation carriers (n = 203). The previous occurrence of colorectal cancer and a family history of SBC did not increase the risk significantly.
Conclusions: Approximately, one out of 25 mutation carriers will develop SBC during life. No specific risk factors were identified. The risk appeared to be too low to advise screening by means of an invasive burdensome procedure like double balloon enteroscopy. However, screening by a non-invasive procedure (videocapsule endoscopy) might be considered if future studies will show its cost effectiveness. In patients with unexplained abdominal complaints and/or unexplained iron deficiency anaemia SBC should be considered.
- CRC, colorectal carcinomas
- DBE, double balloon endoscopy
- GC, gastric cancer
- HNPCC, hereditary non-polyposis colorectal cancer
- LS, Lynch syndrome
- MMR, mismatch repair
- SBC, small bowel cancer
- VCE, video capsule endoscopy
- hereditary nonpolyposis colorectal cancer
- Lynch syndrome
- small bowel cancer
Statistics from Altmetric.com
- CRC, colorectal carcinomas
- DBE, double balloon endoscopy
- GC, gastric cancer
- HNPCC, hereditary non-polyposis colorectal cancer
- LS, Lynch syndrome
- MMR, mismatch repair
- SBC, small bowel cancer
- VCE, video capsule endoscopy
Lynch syndrome (LS) (hereditary non-polyposis colorectal cancer, HNPCC) is defined as an autosomal dominantly inherited disorder of cancer susceptibility. It is characterised by the development of predominantly right sided colorectal carcinomas (CRC) at an early age, and an excess of synchronous or metachronous colorectal carcinomas and extracolonic malignancies.1–4 The condition is caused by a germ-line mutation in one of the DNA mismatch repair (MMR) genes. To date four MMR genes have been found to cause LS: MLH1, MSH2, PMS2, and MSH6. Of these, the MLH1 and MSH2 mutations account for 70–90% of the families with LS.5,6,7,8,9,10,11
Endometrial cancer is the most common site for extracolonic malignancies in LS patients. It has even been reported that the incidence of endometrial carcinoma surpasses the incidence of CRC in female LS patients. Another common extracolonic cancer in LS patients is small bowel cancer (SBC).2,12–16 The location in the small bowel is striking because SBC is very rare in the general population. In Europe fewer then 1 per 100 000 cases are diagnosed each year, and SBC accounts for less then 5% of all gastrointestinal malignancies.17–20 Few studies have been conducted into the lifetime risk for LS patients to develop SBC. The lifetime risks reported in those studies ranged from 1% to 4%, with a relative risk of over 100. A fundamental problem in these studies, however, is that they only report on a small number of LS patients.14–16,21
Even though the relative risk of developing SBC is extremely high, the reported lifetime risk remains relatively low and until recently the possibilities for visualising the small bowel were not very accurate. Therefore screening has always been advised against.16 However, owing to the development of new techniques for visualisation of the small intestine in recent years the question whether screening might be useful has been raised again.18,22
Recently, a paper describing a large number of LS related SBCs was published.22 In this paper it was suggested that, since almost 50% of the SBCs are duodenal carcinomas, screening for proximal SBCs by means of duodenoscopy or push enteroscopy might be beneficial for early detection. However, though the paper gives a useful insight into the characteristics of LS related SBCs, the lifetime risk for LS patients to develop a SBC was not calculated, nor considered in this advice.
To advise on screening for SBC it is paramount that the lifetime risk on SBC is known and taken into account. The aims of the present study were (1) to calculate the lifetime risk for developing a SBC for a large number of known LS patients, and (2) to identify risk factors for SBC useful in the clinical practice to identify patients that might benefit from screening.
PATIENTS AND METHODS
The Dutch HNPCC/Lynch syndrome family registry
Data on patients diagnosed with LS were collected from the Dutch HNPCC registry. This national registry was established in 1987 and aims to promote surveillance and to guarantee follow-up examinations of LS families. The methods and approach of the registry have been described elsewhere.23 In brief, families with clustering of CRC, suspected of LS are referred to the registry from all parts of the Netherlands. Social workers or clinical geneticists trace the pedigree back and laterally as far as possible. The data collected contain personal and medical data of both affected and unaffected family members. The cancer diagnoses are verified by either medical or pathological reports.
Family data
As of January 2006 the registry has collected data on 355 families, 189 of which have a known MMR gene mutation. Only proven or putative MMR mutation carriers were selected for this study. Patients were considered to be a MMR mutation carrier if they had tested positive for an MMR gene mutation or if they were an obligatory carrier because of their position in the pedigree. Putative mutation carriers are those patients diagnosed with CRC before the age of 60 from a LS family with a known mutation. Family members in whom critical data was missing (date of birth, date of death, sex) were excluded from the study. A total of 1496 family members from 189 families met one of the selection criteria.
Risk analysis
The cumulative risk for developing an SBC was calculated by means of a Kaplan-Meier survival analysis, starting from the birth of the patient until an event was recorded. An event was recorded when either the patient developed an SBC, the patient died, or the end date of the study was reached (1 January 2006).
Only those SBC confirmed by medical or pathological report were included in the calculations. Differences in survival for sexes and mutation type were tested for statistical significance by means of a log rank test. An odds ratio was calculated to compare the risks on developing a SBC between families with a case of SBC and those without SBC. An odds ratio was also calculated to evaluate whether the risk for SBC was increased in carriers who had previously been diagnosed with CRC.
RESULTS
Thirty SBCs were diagnosed in 28 (1.9%) of 1496 members from 25 (13%) of 189 LS families. Population characteristics are shown in table 1 and SBC patient characteristics are shown in table 2. In 16 of 28 patients, detailed information was available on the presenting signs or symptoms. In nine cases, the patient presented with unexplained anaemia, six patients presented with small bowel obstruction, and five with abdominal pain. Jaundice, gastrointestinal blood loss, and weight loss were the presenting sign or symptom in one patient each. Thirteen of the 28 SBC patients died before the end date of the study had been reached. The median survival was 6 years (range 0–37 years). Two patients developed a second primary SBC, for both patients the recurrent SBC was in a different part of the small bowel. Thirteen out of the 30 diagnosed SBCs were located in the duodenum (43%), 10 were located in the jejunum (33%), and two in the ileum (7%). The location of five of the SBCs was not reported. Detailed information about the stage of the SBC was reported in 17 of 30 tumours. The tumours included three Dukes A, eight Dukes B, four Dukes C, and two Dukes D tumours.
Characteristics of study population
Characteristics of patients with small bowel cancer
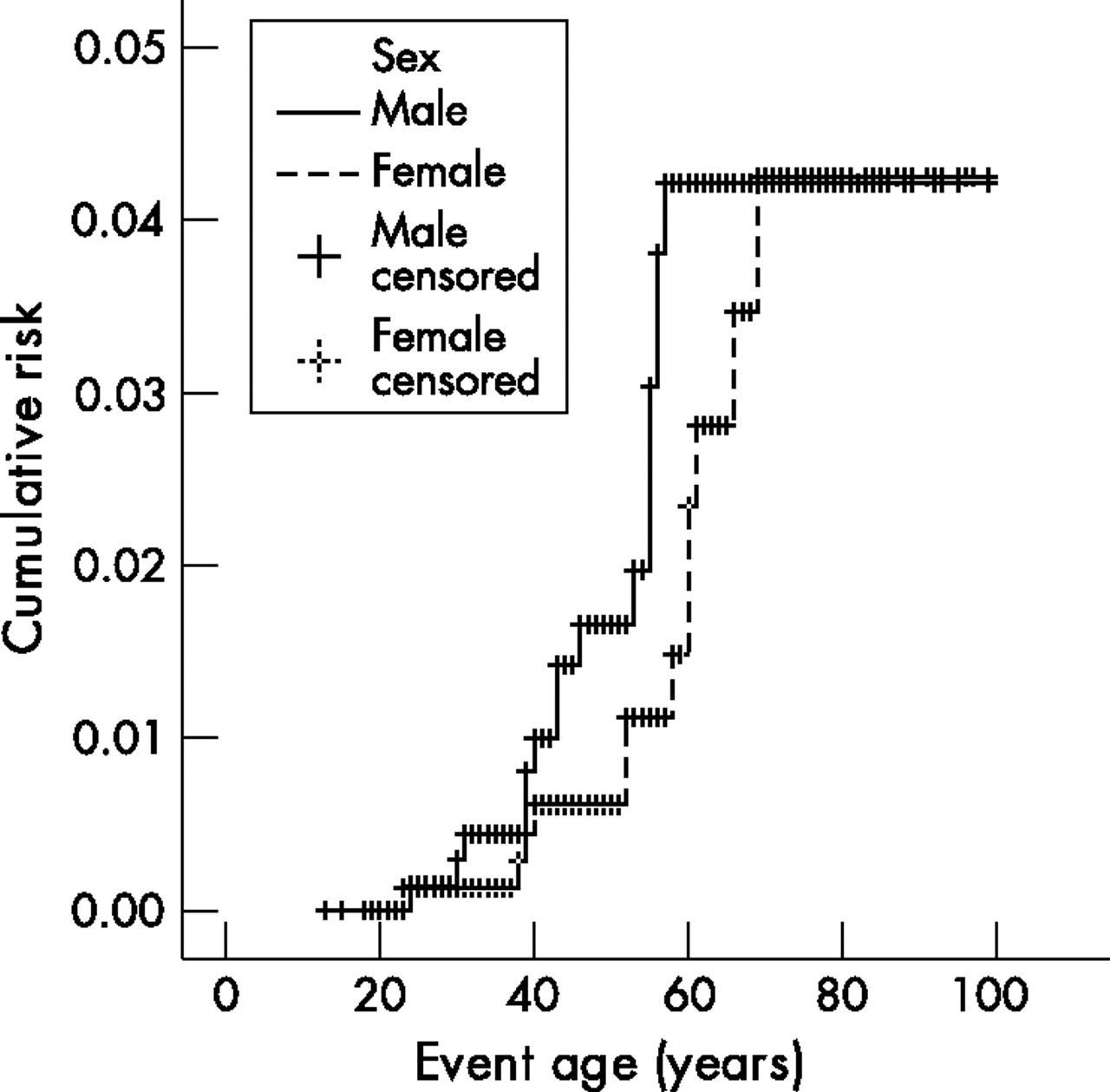
The cumulative risk of developing a SBC was 4.2% (fig 1). The cumulative risks were shown to be the same for both sexes; 4.1% for both male and female MMR mutation carriers (log rank p = 0.2470; fig 2). No SBCs were seen in the MSH6 mutation families. The cumulative risks for MLH1 and MSH2 mutation carriers were shown not to be significantly different; 4.4% and 5.9%, respectively (log rank p = 0.2754; fig 3). Only those family members with known or assumed mutations were included in this study. Of one family the test report on MMR mutation did not state the mutation type. To date we have been unable to retrieve the mutation type. The family has only been excluded for the calculations on mutation type. Since it only concerns one family with five known mutation carriers and no SBC cases, it is not expected to have any relevant effect on the outcome of the calculations.

Lifetime risk of developing small bowel cancer in MMR mutation carriers.

Lifetime risk of developing small bowel cancer in male and female MMR mutation carriers (log rank: p = 0.25).

{kind=link}
{kind=link}
{kind=link}
Lifetime risk of developing small bowel cancer in MLH1 and MSH2 MMR mutation carriers (log rank: p = 0.27).
Thirteen of the 28 patients (46%) were diagnosed with CRC before they were diagnosed with SBC and five of the patients (18%) developed CRC after the diagnosis of SBC. The other 10 SBC patients have not developed CRC to date. For people with a history of CRC the odds ratio for developing a SBC was 1.71 (95% confidence interval 0.81 to 3.63).
Twenty-two families included one case of SBC and three families had two SBC patients each. The odds ratio for the development of a SBC was 1.04 (95% CI 0.42 to 2.60) for LS patients with a SBC in the family.
DISCUSSION
The generally accepted World Health Organization criteria for screening require that the risk of developing a specific cancer in the target group is known. Secondly, data on the natural history, an acceptable screening test with high sensitivity and specificity and curative treatment should be available. Finally, the prognosis should be influenced favourably by early treatment, and the costs of the surveillance programme should be acceptable.16 The current paper aimed to assess the first two criteria for screening programmes and to give valid advice on screening LS patients for SBC.
Though the relative risk reported for small bowel carcinomas in LS families is extremely high, the present study showed that the absolute lifetime risk for MMR mutation carriers to develop an SBC was only 4.2%. Almost 50% of these tumours were found in the duodenum with a clearly decreasing frequency from the duodenum to the ileum. The investigated risk factors sex, family history, mutation type, and CRC were not found to significantly increase the lifetime risk.
The current study calculated the cumulative risk in the largest cohort of LS patients from families with a known MMR defect so far reported. All SBCs reported were confirmed by either medical or pathological report. The cumulative risk for the development of an SBC agrees with those reported by others.14–16
It was shown that the cumulative risk for male and female HNPCC patients was similar. Earlier studies showed a higher frequency of SBC in males compared to females. However the cumulative risk for both sexes has not been reported previously.22,24,25
All of the SBCs were identified in members of families with a mutation in MLH1 or MSH2. However SBCs families with a mutation in MSH6 or PMS2 have been reported in the literature. The relative small proportion of families with MSH6 mutations in the present study might be the explanation for finding SBC only in carriers of a mutation in MLH1 or MSH2. No significant difference in cumulative risk was found between MLH1 and MSH2 families. Earlier studies also did not show a significant difference in occurrence of SBC between MSH1, MLH2, MLH6, and PMS2 families.7,22,24,26
Thirteen of the patients had a history of CRC before they were diagnosed with SBC. The previous occurrence of CRC did not lead to a significantly increased risk for developing a SBC. Other studies showed that more often SBC was the first clinical manifestation of HNPCC.22,24
Altogether, we identified 28 patients with SBC in 25 families. A maximum number of two SBC cases per family was observed, which suggests that there is no significant familial clustering of SBC. Moreover, having a family member with SBC was not correlated with an increased risk for developing SBC. The family clustering of SBC has not been extensively investigated previously, but none of the studies reported evidence for familial clustering.21,22,24
What are the implications of these results on the recommendations for screening on SBC? Previous studies have always advised against screening, because of the relatively low risk and lack of sensitive imaging tools.16 Recent years have seen the development of new techniques such as video capsule endoscopy (VCE) and double balloon endoscopy (DBE). The development of these tools led to the question whether a screening programme with one or more of the new tools would be worthwhile in LS patients.
DBE is known to be a reliable tool for the detection of lesions in the small bowel and allows for direct access to the lesion. However, DBE is a relatively invasive time consuming burdensome procedure and, therefore, periodic screening by means of DBE appears not to be justified for these low risk tumours.
VCE is a promising new technology, which is non-invasive and has proved its usefulness for the detection of small bowel lesions in familial adenomatous polyposis and Peutz Jeghers patients and for the evaluation of occult gastrointestinal bleeding.27–31 VCE has been reported to be superior to magnetic resonance imaging of the small bowel for the identification of polyps less than 15 mm in familial adenomatous polyposis and Peutz Jeghers patients.32 However, to date no study has been performed into the possibility for screening and the cost effectiveness of screening LS patients by means of a VCE.
In a recent paper by the German HNPCC consortium, the authors proposed regular screening for gastric cancer (GC) and proximal SBC in LS patients by gastroduodenoscopy.27 Since approximately 50% of the SBCs would be in reach of the gastroduodenoscope, the cost effectiveness will also depend on the lifetime risk on GC. The German consortium reported that GC was the third most common cancer in German LS patients; however, the lifetime risk on GC was not calculated.27 In the Finnish LS population it was found that the lifetime risk on GC was high, over 10%.12 However, a comparison of Dutch with Korean LS patients showed that international figures can not be readily translated to a specific country.33 A Dutch study showed a low relative risk of developing GC in LS patients.16 This makes it unlikely that a GC screening programme using gastroduodenoscopy would be cost effective in the Netherlands. However, before a final statement on the value of surveillance by gastroduodenoscopy can be made, further evaluation of the combined risk of GC and proximal SBC has to be performed.
In conclusion, the present study demonstrated that one out of 25 germline mismatch repair gene mutation carriers will develop SBC during life. The risk appeared to be too low to advise screening by means of an invasive burdensome procedure like DBE. Screening by a non-invasive procedure (VCE) might be considered if future studies show its cost effectiveness. Because of the significant risk for LS patients developing SBC, SBC should be considered and excluded in all patients from LS families with unexplained abdominal complaints and/or unexplained iron deficiency anaemia. Although the present study did not prove that such a policy will lead to an early diagnosis, as a general rule in patients with malignancies it is plausible that a prompt diagnosis and treatment will improve the prognosis.
REFERENCES
Footnotes
-
Published Online First 4 April 2007
Linked Articles
- Digest