Article Text

Abstract
Background: The rate of progression to cirrhosis varies among individuals chronically infected with the hepatitis C virus (HCV). Coagulation pathway activation in models of hepatic fibrosis suggests variation in coagulation pathway components may influence the rate of fibrosis. We hypothesised that polymorphisms of the coagulation factors II and V affect the rate of progression to cirrhosis in HCV infected subjects.
Methods: We studied the relationship between rate of fibrosis (calculated by dividing the fibrosis stage by duration of infection) and genotypes of specific coagulation pathway genes in 352 White European patients infected with HCV. Genotyping was performed using reverse line blot hybridisation.
Results: The rate of fibrosis was significantly higher in patients with the factor V Leiden genotype (Arg560Gln) (ANOVA, p=0.004). In disease association studies, a significant association was seen (Fisher’s exact test, p=0.029; odds ratio 3.28 for fast progression to cirrhosis (expected to reach cirrhosis in less than 30 years) if heterozygous for factor V Leiden). No associations were seen between factor II genotype and fibrosis rate.
Conclusions: Possession of the factor V Leiden polymorphism significantly increases the risk of rapid disease progression in HCV, suggesting a role for the coagulation system in the pathogenesis of fibrotic liver disease.
- hepatitis C
- fibrosis
- rate
- factor V Leiden
- polymorphism
- HCV, hepatitis C virus
- PCR, polymerase chain reaction
- ANOVA, analysis of variance
- ANCOVA, analysis of covariance
Statistics from Altmetric.com
- HCV, hepatitis C virus
- PCR, polymerase chain reaction
- ANOVA, analysis of variance
- ANCOVA, analysis of covariance
Hepatitis C virus (HCV) infects 170 million people worldwide, and the majority of the morbidity and mortality associated with infection is due to the development of cirrhosis and its complications. The rate of progression to cirrhosis through the deposition of fibrous tissue varies among individuals. Only a small proportion of the variability in the rate of fibrosis can be explained by known demographic and environmental factors (18% in one large study1 and 29% in another2).
There are a number of both general and specific lines of evidence to suggest that host genetic factors play a key role. Host genotype has been demonstrated to determine outcome in a number of infectious diseases—for example, malaria,3 hepatitis B virus,4,5 and HCV.6 Previous work in hepatitis C has examined the role of the HLA system in spontaneous clearance of the virus6,7 and the role of transforming growth factor β1 in conjunction with the renin angiotensin system on fibrosis stage.8 There is controversy over the impact of HFE polymorphisms on the outcome of HCV infection.9,10 The natural history of HCV has been shown to vary substantially even where age group, sex, and viral variables are controlled, as demonstrated in a cohort of Irish women infected through contaminated anti D (RHO) immunoglobulin.11 Identification of specific genetic factors may assist in prognosis, therapeutic decisions, and even therapeutic discovery.
The coagulation pathways are of importance in liver injury as elsewhere in the body, with supporting evidence from carbon tetrachloride induced fibrosis in the rat demonstrating increased fibrin and fibrinogen deposition in the injured liver.12 Furthermore, thrombin receptors on stellate cells are upregulated during liver damage13 and thrombin is also known to be a stellate cell mitogen.14 A number of coagulation factor polymorphisms have been shown to have functional effects on the clotting cascade and subsequent risk of thrombosis. An adenine for guanine substitution at position 20210 in the 3′ untranslated region of the thrombin gene is associated with excess thrombin generation15 and an increased risk of both arterial and venous thrombosis. Factor V Lieden results from an amino acid substitution at position 506 of Gln for Arg and follows a single nucleotide substitution (adenine for guanine at position 1691). The polymorphism confers resistance to activated protein C, part of a negative feedback loop which inhibits activated factor V. Together with activated factor X, factor V converts prothrombin to thrombin. The factor V Leiden mutation has been shown to increase the risk of venous thromboembolism.16 We examined the hypothesis that the procoagulant tendency conferred by these polymorphisms leads to increased deposition of fibrous tissue and hence a more rapid progression towards cirrhosis.
METHODS
Patients
Patients with chronic HCV infection seen at eight European hepatology centres17 who had undergone at least one liver biopsy as part of routine clinical practice between 1 January 1990 and 30 June 2001 were identified. All patients were white Europeans known to be HCV antibody positive and all were known to be RNA positive. Patients were excluded if they had been given antiviral therapy prior to biopsy, if they had hepatocellular carcinoma, evidence of other types of liver disease in addition to their hepatitis C, human immunodeficiency virus coinfection, or a non-interpretable biopsy. Data were collected with regard to patient demographic details (sex, date of birth, age at infection, risk factor, ethnic origin), histological parameters, and viral genotype. Date of infection was documented; in those infected by blood products, the date of transfusion was recorded. When the exact date was not known, but the year was, the estimated date of infection was recorded as the middle of that year. In patients infected by intravenous drug use, the date of infection was estimated as the middle of the year of first drug use, as elsewhere.1 Patients for whom date of infection was unknown were excluded.
Histopathology
Biopsies from all patients were stained with both haematoxylin-eosin and reticulin. Scoring was by a single pathologist (RG) using the modified histological activity index (HAI- Ishak) scoring system.18 This scoring system assigns a score of 0–6 for the degree of fibrosis. Fibrosis stage was assumed to be zero at the time of infection.
DNA collection
All patients gave informed consent. The local regional ethics committees of each centre gave approval for the study. Genomic DNA was extracted from a 5 ml sample of whole blood collected into EDTA using a commercial kit (Nucleon II, Scotlabs, UK).
Genotyping
Genotyping was performed by a reverse line blot hybridisation linear array assay (Roche Molecular Systems, Alameda, USA). Extracted genomic DNA was subjected to multi-target polymerase chain reaction (PCR) amplifications using a blend of primers. PCR products were then genotyped by hybridising to sequence specific oligonucleotides impregnated onto nylon strips and detected in a colorimetric reaction, as described by Cheng and colleagues.19 Strips were then examined for the presence of coloured bands indicating the presence of an allele.
Statistical analysis
All statistics were performed using SPSSv10 (SPSS inc, Chicago, Illinois, USA) and Microsoft Excel. Rate of fibrosis was calculated as the ratio of fibrosis score to duration of infection at biopsy. Median rate of fibrosis for the cohort was determined and patients with rates either side were designated “fast” or “slow” fibrosers, accordingly. Demographic differences between the fast subgroup (predicted cirrhosis in less than the median) and the slow subgroup (not predicted to reach cirrhosis for more than the median) were assessed using independent sample t tests, analysis of variance (ANOVA), and χ2 tests, as appropriate. Rate of fibrosis was logarithmically transformed to give a normal distribution. Comparison of rates between individuals with different genotypes was performed using ANOVA. Allele frequencies were compared between subsets of patients with fast or slow fibrosis using the χ2 or Fisher’s exact test, as appropriate. A sensitivity analysis, taking different cut off points between “fast” and “slow” fibrosers, was also performed.
Variations in rates of fibrosis with respect to genotype were sought. In addition, genotype and allele frequencies were compared between fast and slow subgroups of this cohort. In order to account for demographic differences between the phenotypic groups, subgroup analysis was performed by sex and analysis of covariance (ANCOVA) to examine the effect of age at infection, necroinflammatory score, and sex.
Comparison of fibrosis stage between genotypes was performed after correction for duration of infection using ANCOVA.
RESULTS
Reproducibility of fibrosis assessments.
There was good intra and interobserver agreement (weighted kappa 0.87 and 0.82, respectively) on a random sample of 35 biopsies.
Demographic features
Median rate of fibrosis was 0.2 fibrosis units per year (30 years to cirrhosis). Demographic features for the groups of patients with “fast” and “slow” fibrosis are shown together with those for the group as a whole in table 1. As expected, there was an excess of males in the fast fibrosis group (p=0.001) and age at infection was significantly higher in those with rapid fibrosis (p<0.001). In this group, mode of acquisition of HCV and viral genotype were not related to rate of fibrosis progression. Fast fibrosers had a shorter duration of infection prior to biopsy and this correlated inversely with age at infection (r=−0.485; p<0.001). The fast fibrosis group had a median rate of fibrosis fivefold greater than the slow fibrosis group (0.4 units/year v 0.08 units per year; p<0.001). Mean fibrosis score and necroinflammatory score were both higher in the fast fibrosis group (3.67 and 5.15 units v 1.68 and 3.7 units; p<0.001) (table 1).
Demographic differences between the fast and slow fibrosis groups
Comparison of fibrosis rates
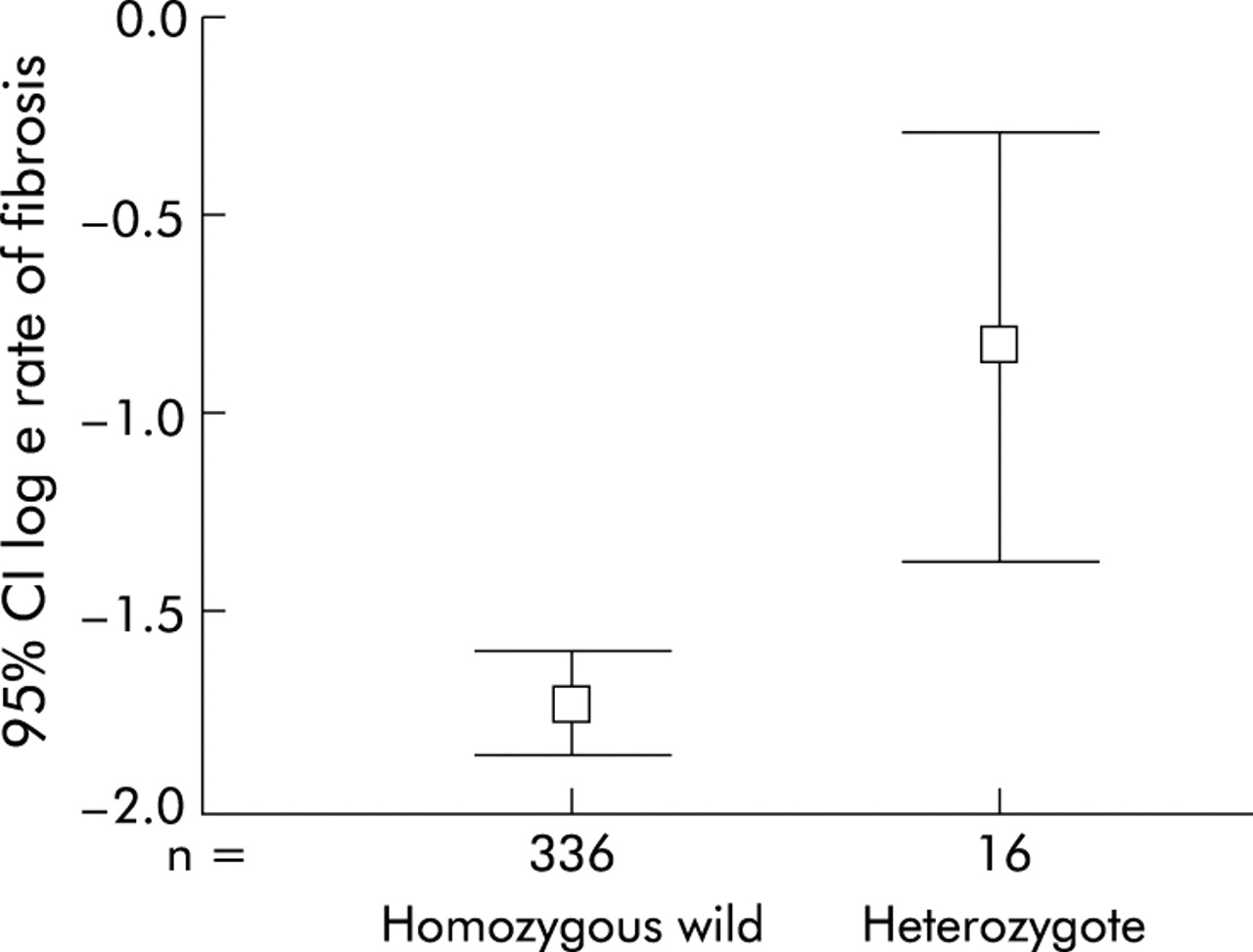
The relationship between fibrosis rate and genotype was examined using ANOVA (rate of fibrosis was logarithmically transformed to achieve a normal distribution). A total of 352 individuals genotyped for factor V Leiden were included (table 2). There was a significant influence of factor V genotype on the rate of fibrosis. Median rate of fibrosis in patients who were heterozygous for the factor V Leiden mutation was 0.37 fibrosis units per year (interquartile range 0.2–0.89) compared with 0.18 fibrosis units per year (interquartile range 0.08–0.37) in patients with wild-type factor V (ANOVA, p=0.004) (fig 1). No patient was homozygous for factor V Leiden.
Summary of disease association and ANOVA results for factor V Leiden

Rate of fibrosis by genotype: 95% confidence intervals for loge rate of fibrosis by factor V Leiden genotype (ANOVA, p=0.004).
Disease associations
Disease associations were performed with DNA from 182 individuals with slow fibrosis rates and 170 with fast rates. A significant association was seen (Fisher’s exact test, p=0.029; odds ratio 3.28 (95% confidence interval 1–12.7) for fast v slow) (table 2).
Sensitivity analysis
Disease associations for different cut off points defining fast and slow fibrosers were performed (table 3). The more extreme the differences in rate of fibrosis, the greater the likelihood of an individual possessing the factor V Leiden allele. For example, patients progressing to cirrhosis in less than 15 years were 12.3 time more likely to have the factor V Leiden allele than those progressing in over 35 years (p=0.0005).
Sensitivity analysis showing strength of the association between possession of factor V Leiden and different cut offs for “fast and slow”
Multivariate analysis
The effect of factor V genotype on the rate of fibrosis was most pronounced in males (p=0.007, Fisher’s exact test; odds ratio 10 for fast v slow fibrosis, ANOVA, p=0.021).
To take into account the influence of age at infection, necroinflammatory score, and sex on rate of fibrosis, ANCOVA using a type 1 sum of squares model was performed. The significant effect of factor V genotype on rate of fibrosis progression was sustained (p=0.007) when age at infection (p<0.001), necroinflammatory score (p<0.001), and sex (p<0.001) were taken into account.
Fibrosis stage and possession of factor V Leiden
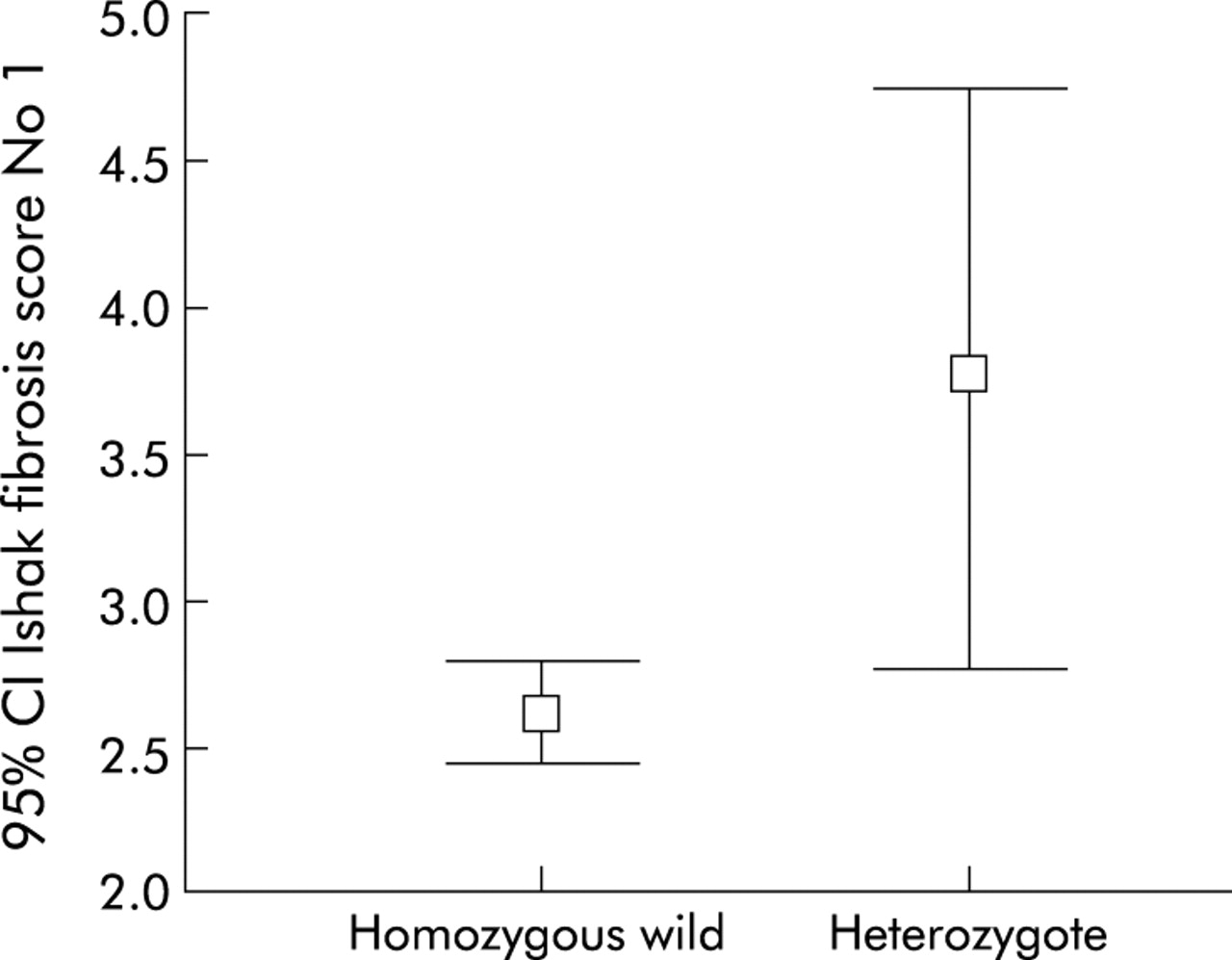
Frequencies of factor V genotypes for patients at different fibrosis stages are shown in table 4 and fig 2. Heterozygotes for factor V Leiden had a mean fibrosis score of 3.75 units compared with 2.61 units for homozygous wild-type patients (p=0.01). This difference was sustained after correction for duration of infection (p=0.011).
Frequencies of different factor V genotypes in patients at different stages of fibrosis

{kind=link}
{kind=link}
Stage of fibrosis according to factor V genotype: 95% confidence intervals for stage of fibrosis by factor V Leiden genotype (ANOVA, p=0.01).
Factor II
No significant association was seen between factor II genotype and rate of fibrosis. In the fast group there were 112 homozygous wild-types and six heterozygotes compared with 175 and seven, respectively, in the slow group (odds ratio 1.3; p=0.6, χ2; p=0.59, ANOVA).
DISCUSSION
These data provide evidence that possession of the factor V Leiden mutation leads to an increased rate of fibrosis in HCV infection. The association of rapid fibrosis with the FvL mutation was independent of potential confounding variables and the magnitude of effect was more pronounced in males than in females. The use of rate of fibrosis in HCV as a disease association phenotype is novel. The use of the metric rate of fibrosis is not universally accepted as it implies that the time between fibrosis stages is equal. Fibrosis stage is a morphological and possibly a semiquantitative variable. However, in a cross sectional analysis of patient cohorts, including our own,1,2 there was a linear relationship between duration of infection and fibrosis stage. Moreover, fibrosis stage in a population increases linearly as the population ages. This has proved to be a reliable means of comparing disease progression in cohorts as opposed to individual patients. If this were not the case, and there was more rapid transit through higher stages of fibrosis, our findings are still valid as we have shown that the fibrosis score in our rapid fibrosis group was significantly higher despite a shorter duration between infection and the first biopsy. In table 4 and fig 2, we demonstrated that there was also an association between possession of factor V Leiden and a more advanced stage of fibrosis. This remains after allowance for duration of infection.
The functional significance of factor V Leiden is well described in that this mutation confers resistance to activated protein C which normally degrades factor V. This finding suggests that liver inflammation resulting from HCV infection leads to activation of the coagulation system. In those patients with the factor V Leiden mutation, the degree of activation is enhanced leading to increased thrombin activity and fibrin production. Thrombin is a stellate cell mitogen14 and therefore activation of the coagulation cascade may stimulate stellate cell activation and fibrosis. Factor II polymorphisms occur at lower frequency than factor V and we are unable to exclude a type 2 error in this population. However, the data indicate a weak trend between the procoagulant heterozygote form and fast fibrosis in the expected direction. The factor V Leiden polymorphism affects the function of the molecule whereas the thrombin polymorphism affects plasma levels, implying that there may be additional ways in which it exerts its effect—possibly even independently of the clotting cascade.
A link between coagulation and fibrosis has now been demonstrated in a transgenic factor V Leiden mouse model20 with increased fibrin deposition in all tissues. Moreover, pulmonary fibrosis in response to bleomycin is significantly increased in these animals.21 The association of factor V Lieden with rapid development of liver fibrosis appears to be predominant in males with only a trend observed in females. As has been previously reported by Poynard and colleagues,1 age at infection and male sex were found to be associated with a rapid fibrosis rate in our study. Fibrosis has considerable pathological similarity to atherosclerosis which is also more common in males. It may be that oestrogens protect women from fibrosis in a similar way that premenopausal women are protected against ischaemic heart disease. The lack of association between alcohol and rate of fibrosis in our population is surprising given the findings of Poynard1 and may relate to different drinking patterns in our population. It is possible that in our patient group admission of alcohol abuse was considered taboo leading to misrepresentation of genuine consumption, particularly as the data were collected after the patient became aware of their HCV diagnosis. Data collection in this area is notoriously inaccurate and approximately half of the cohort reported abstinence while some did not respond at all. Selective recall by those with more severe disease could give an association with severity without demonstrating an effect on rate. We were only able to record current alcohol intake and patients aware of their diagnosis may have modified their drinking behaviour in accordance with medical advice. However, it is difficult to hypothesise how alcohol intake and factor V Leiden genotype may interact to produce a confounding effect. Viral genotype was only available for approximately 60% of the cohort and we did not demonstrate any effect on the rate of fibrosis. For both alcohol and viral genotype, there was an equal proportion of missing data in both the slow and fast fibrosis groups, reducing the possibility of any bias.
It is possible that factor V Leiden may influence the rate of fibrosis in hepatitis of other aetiologies but we were unable to address this hypothesis in this study. Many factors affect the rate of development of fibrosis. Individual genes play only a small part in the overall variability between individuals. Clearly possession of factor V Leiden is neither a necessary nor a sufficient cause of rapid fibrosis and this is demonstrated by the finding that even in the fast fibrosis group it accounted for only a minority of cases. However, these data may implicate a general involvement of coagulation in the fibrosis pathway, with those who have a procoagulant tendency also having a profibrotic one.
Acknowledgments
The reverse line blot hybridisation system was kindly donated by Roche Molecular Systems, Inc, Alameda, California, 94501 USA.
HENCORE is the Hepatitis C European Network for Cooperative Research. The participants were: Pierre Pradat PhD (Hopitaux de Lyon, Place de l’hopital, 69288 Lyon, France); Juan Esteban MD (Hospital General Valle hebron, Pasea Valle, Hebron S/U o8035, Barcelona, Spain); Stephanos Hadziyannis MD (University of Athens Medical School, 2nd Department of Medicine, Hippokration Hospital, 114 Vasilissis Sophias Avenue, Athens 610, Greece); Michael Manns PhD (Department Gastroenterology, Molinette Hospital, Torino, Italy); Hans Tillmann MD (Medizinische Hochschule Hannover, Carl Neuburg Str 1, 30623 Hannover, Germany); Alfredo Alberti MD, Liliana Chemello MD (Department of Clinical and Experimental Medicine, Via Guistiniani 2, 35126 Padova, Italy); Giorgio Saracco MD, Mario Rizzetto PhD (Department of Gastroenterology, Molinette Hospital, Torino, Italy); Jean-Henrik Braconier MD (Department of Infectious Diseases, University Hospital Lundt, Sweden); Christian Trepo (Hopitaux de Lyon, 1 Place de l’Hopital, 69288 Lyon Cedex 02, France).
Part of this work was generously supported by funds donated by Roche Discovery, Welwyn, UK and the Charmot-Horton Foundation. The corresponding author is a Medical Research Council of Great Britain clinical training fellow.