Article Text

Abstract
Background and aims: Many lines of evidence suggest that T helper cell type 1 (Th1) immune responses predominate in Crohn’s disease (CD). Recently, a novel transcription factor T-box expressed in T cells (T-bet) has been reported as the master regulator of Th1 development. This study was designed to investigate the role of T-bet and proinflammatory cytokines in Th1 mediated immunopathology in CD.
Materials: CD4+ lamina propria mononuclear cells (LPMCs) were isolated from surgically resected specimens (CD, n = 10; ulcerative colitis (UC), n = 10; normal controls (NL), n = 5).
Methods: (1) T-bet expression of CD4+ LPMCs was examined by quantitative real time polymerase chain reaction and western blotting. (2) T-bet expression of LPMCs stimulated by interleukin (IL)-12/IL-18 was analysed by western blotting. (3) Interferon γ (IFN-γ) production and T-bet expression of CD4+ peripheral blood mononuclear cells (PBMCs) were examined with or without stimulation by anti-CD3/CD28 monoclonal antibodies and/or IL-12.
Results: (1) T-bet expression of CD4+ LPMCs was increased in CD compared with UC and NL. (2) Synergistically, augmentation of IFN-γ production by IL-12/IL-18 was independent of T-bet expression in LPMCs. (3) T-bet was induced by T cell receptor stimulation in CD4+ PBMCs. T-bet induction correlated with IFN-γ production and with augmentation of surface expressed IL-12 receptor β2.
Conclusions: T-bet induction by antigenic stimulation and subsequent stimulation by macrophage derived IL-12/IL-18 are important for establishing Th1 mediated immunopathology in CD.
- CD, Crohn’s disease
- UC, ulcerative colitis
- NL, normal control
- IL, interleukin
- IL-12Rβ2, IL-12 receptor β2
- LPMC, lamina propria mononuclear cell
- PBMC, peripheral blood mononuclear cell
- T-bet, T-box expressed in T cells
- TCR, T cell receptor
- Th1, T helper cell type 1
- IFN-γ, interferon γ
- mAb, monoclonal antibody
- TNF-α, tumour necrosis factor α
- PCR, polymerase chain reaction
- AU, arbitrary units
- TGF-β, transforming growth factor β
- T-bet
- Crohn’s disease
- ulcerative colitis
- interleukin 12
- interleukin 18
Statistics from Altmetric.com
- CD, Crohn’s disease
- UC, ulcerative colitis
- NL, normal control
- IL, interleukin
- IL-12Rβ2, IL-12 receptor β2
- LPMC, lamina propria mononuclear cell
- PBMC, peripheral blood mononuclear cell
- T-bet, T-box expressed in T cells
- TCR, T cell receptor
- Th1, T helper cell type 1
- IFN-γ, interferon γ
- mAb, monoclonal antibody
- TNF-α, tumour necrosis factor α
- PCR, polymerase chain reaction
- AU, arbitrary units
- TGF-β, transforming growth factor β
Crohn’s disease (CD) is a chronic inflammatory process involving the gastrointestinal tract, characterised by discontinuous and transmural inflammation. Although the aetiology of CD is not fully understood, accumulating evidence suggests that dysregulation of the local immune system is pivotal in the pathogenesis of CD.1,2 Studies from humans and experimental murine colitis models indicate that in CD, the local immune response tends to be predominantly T helper cell type 1 (Th1) and is reflected by local release of cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-12, and IL-18.3–7 Studies conducted in experimental murine colitis models showing that neutralising antibodies against TNF-α, IL-12, or IL-18 prevent the onset of colitis lend further support to a Th1 predominant immunopathology in CD.8–10 Indeed, antihuman TNF-α monoclonal antibody (mAb) (Infliximab) is effective for many patients with CD refractory to conventional therapy.11,12 Collectively, these observations indicate that Th1 mediated immunopathology plays a central role in induction and perpetuation of intestinal inflammation in CD.
Given the observed polarised nature of T cells in CD, understanding the mechanisms that lead to the establishment of this polarised state in the gastrointestinal mucosa is critical. Naïve CD4 T lymphocytes in transit to becoming either Th1 or Th2 effector cells undergo sequential stages of cytokine activation, commitment, silencing, and physical stabilisation during polarisation into differentiated effector subsets, a process tightly controlled by regulatory transcription factors.13,14 Various transcription factors such as c-maf, GATA-3, and STAT-6 have been shown to promote expression of several Th2 cytokines, including IL-4, IL-5, and IL-13, either by transactivation of cytokine gene promoters and enhancers or induction of chromatin remodelling.15–17 In contrast with Th2 differentiation, very little is known about the molecular basis of Th1 differentiation. STAT-4 and IRF-1 are specifically associated with IL-12 and interferon (IFN)-γ signalling in T cells, respectively, and play a key role in regulating cytokine production of Th1 cells at the transcriptional level.13,18
Recently, T box expressed in T cells (T-bet), a member of the T-box family of transcription factors, has been shown to transactivate the IFN-γ gene.19 T-bet, whose expression is primarily limited to the immune system, is rapidly induced in early developing Th1 cells and is absent in developing Th2 cells.19 T-bet deficient mice show normal lymphoid development but exhibit marked impairment in mounting Th1 mediated immune responses in response to IL-12.20 Moreover, retroviral mediated expression of T-bet in Th2 cells leads to induction of a Th1 cytokine profile in these cells.19 Thus T-bet initiates Th1 cell differentiation by activating Th1 genetic programmes and repressing Th2 programmes.
Neurath et al have demonstrated that expression of T-bet is increased in CD and that T-bet regulates the mucosal cytokine balance in various murine experimental colitis models.21 However, the pathophysiological role of T-bet in human CD has not been fully described. In the present study, we investigated the role of T-bet in induction of Th1 responses in human CD, and differences in immune responses between CD and ulcerative colitis (UC).
MATERIALS AND METHODS
Patients and samples
Mucosal samples were obtained from surgically resected inflamed areas of intestinal specimens from 10 patients with CD and 10 with UC. Patient profiles are summarised in table 1. CD or UC was diagnosed based on clinical, radiographic, endoscopic, and histological findings by established criteria.22,23 The degree of inflammation was histologically moderate to severe in all samples. Normal controls (NL) included mucosal samples from macroscopically and microscopically unaffected areas from patients with sporadic colon cancer. All experiments were approved by the local ethics committees. Informed consent was obtained from all patients before obtaining the samples.
Clinical profiles of Crohn’s disease (CD) and ulcerative colitis (UC) patients
RNA extraction and quantitative real time polymerase chain reaction (PCR)
Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Hilden, Germany). Total RNA was treated with Qiagen DNase (Qiagen) to remove any contaminating genomic DNA. Complementary DNA (cDNA) was synthesised using the Superscript first strand synthesis system for reverse transcription-PCR (Invitrogen, Carlsbad, California, USA) according to the manufacturer’s instructions. Quantitative real time PCR was performed using SYBR Green PCR master mix (Applied Biosystems, Foster City, California, USA) with an ABI PRISM 7700 Sequence Detection System (Applied Biosystems). We confirmed that non-specific bands were not detected by melting curve analysis for each primer set. T-bet mRNA transcripts were normalised with β-actin mRNA transcripts and expressed as arbitrary units (AU). PCR primers were as follows: T-bet forward, 5′-CCC CCA AGG AAT TGA CAG TTG-3′; reverse 5′-GGG AAA CTA AAG CTC ACA AAC-3′. β-Actin forward, 5′-AAG CAG GAG TAT GAC GAG TCC G-3′; reverse, 5′-CGG AAC TAA GTC ATA GTC CGC C-3′
Preparation of lamina propria mononuclear cells (LPMCs) and peripheral blood mononuclear cells (PBMCs)
LPMCs were isolated from surgically resected intestinal specimens using enzymatic techniques, as previously described.3 Briefly, dissected mucosa was incubated in calcium and magnesium free Hanks’ balanced salt solution (Sigma, St Louis, Missouri, USA) containing 2.5% fetal bovine serum (BioSource, Camarillo, California, USA) and 1 mM dithiothreitol (Sigma). The mucosa was then incubated in medium containing 0.75 mM EDTA (Sigma) for 60 minutes at 37°C. During this treatment, intraepithelial lymphocytes and epithelial cells were removed from the tissue. Then, tissues that contained LPMCs were collected and incubated in medium containing 0.02% collagenase (Worthington Biochemical Corp, Freehold, New Jersey, USA). The fraction was pelleted and cells centrifuged over a 40–60% Percoll solution (Amersham Biosciences Corp, Piscataway, New Jersey, USA) density gradient. PBMCs were isolated by density gradient centrifugation using Lymphoprep (Nycomed Pharma, Oslo, Norway) from heparinised peripheral blood samples. LPMCs or PBMCs were further separated into CD4 positive cells using MACS (Miltenyi Biotec, Auburn, California, USA) according to the manufacturer’s instructions.
Cell culture
LPMCs and CD4+ PBMCs were cultured at a concentration of 1×106/ml in complete RPMI 1640 medium (Sigma) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin (Invitrogen). For CD4+ PBMCs culture, 10 μg/ml of immobilised anti-CD3 (OKT3) and 5 μg/ml of anti-CD28 (CD28.2; BD Pharmingen, San Diego, California, USA) antibodies were used. Recombinant IL-12p70 (BD Pharmingen) and/or IL-18 (MBL, Nagoya, Japan) were added in culture medium, as indicated.
Enzyme linked immunosorbent assay (ELISA)
Concentrations of IFN-γ and IL-12 in culture supernatants of LPMCs and PBMCs were measured using specific ELISA (IFN-γ: Endogen, Woburn, Massachusetts, USA, IL-12: BioSource, Camarillo, California, USA). According to the manufacturer’s instructions, the minimum detectable IL-12 and IFN-γ concentrations were 7.8 pg/ml and 25.6 pg/ml, respectively.
Flow cytometry
Flow cytometric analysis was performed as previously described.9 Phycoerythrin conjugated antihuman IL-12 receptor β2 (IL-12Rβ2) antibody was purchased from BD Pharmingen. Fluorescence intensity on the surface of the cells was analysed using a FACScan (Becton Dickinson, Mountain View, California, USA).
Protein extraction and western blotting
Total protein was extracted using lysis buffer containing 10 mM Tris HCl, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, and a mixture of protease inhibitors. Total protein (10 μg) was separated on a NuPAGE 4–12% Bis-Tris gel (Invitrogen) and electrophoretically transferred onto Immobilon-P membrane (Millipore, Bedford, Massachusetts, USA). To detect T-bet protein, the membrane was incubated with rabbit anti-T-bet antisera (1:3000 final dilution; kindly provided by Drs L Glimcher and S Szabo, Harvard, School of Public Health, Boston, Massachusetts, USA) and subsequently with horseradish peroxidase conjugated goat antirabbit IgG Ab (1:2000, New England Biolabs, Beverly, Massachusetts, USA). Antibody reactions were detected with a chemiluminescence detection kit (Amersham Biosciences Corp). The membrane was subsequently stripped with Restore western blot stripping buffer (Pierce, Rockford, Illinois, USA) and incubated with mouse anti-β-actin Ab (1:2000, Sigma). Densitometric analysis was performed with NIH image software version 1.61 and T-bet expression was adjusted to β-actin expression.
Statistical analysis
Statistical differences were analysed using the Mann-Whitney U test. A p value of <0.05 was considered to be significant. All data are expressed as mean (SEM).
RESULTS
T-bet was upregulated in CD4+ LPMCs of CD
To clarify the involvement of T-bet in Th1 mediated immunopathology in CD, we first assessed expression of T-bet mRNA in CD4+ LPMCs obtained from five NL, 10 CD patients, and 10 UC patients using quantitative real time PCR. T-bet mRNA obtained from CD4+ LPMCs of CD patients (1.59 (0.38) AU) was significantly (p<0.01) increased compared with that obtained from UC patients (0.48 (0.14) AU) and NL (0.41 (0.11) AU) (fig 1A). T-bet protein was also significantly (p<0.05) increased in CD4+ LPMCs of CD patients (88.5 (21.3) AU) compared with those of UC patients (12.7 (7.2) AU) and NL (4.1 (3.1) AU) (fig 1B). T-bet expression was not significantly different between UC and NL at both the mRNA and protein levels.

T-box expressed in T cells (T-bet) expression of CD4+ lamina propria mononuclear cells (LPMCs) was increased in Crohn’s disease (CD). Total RNA was extracted from CD4+ LPMCs (CD, n = 10; ulcerative colitis (UC), n = 10; normal controls (NL), n = 5). (A) Levels of T-bet mRNA transcripts were measured by quantitative real time polymerase chain reaction and adjusted to β-actin mRNA transcripts. **p<0.01. (B) T-bet protein expression was assessed by western blotting in CD and UC patients and in NL. AU, arbitrary units. (C) LPMCs were cultured in medium alone for 48 hours (CD, n = 6; UC, n = 6; NL, n = 6). Production of interferon γ (IFN-γ) was measured by ELISA. **p<0.01.
To assess the correlation between T-bet expression and Th1 responses, we next examined IFN-γ production by LPMCs cultured without any stimuli for 48 hours, as measured by ELISA. As shown in fig 1C, IFN-γ production by LPMCs from patients with CD was significantly (p<0.01) higher (1659.0 (206.7) pg/ml) than that from patients with UC (461.5 (88.1) pg/ml) or from NL (233.3 (61.4) pg/ml). This enhanced production of IFN-γ well corresponded to augmentation of T-bet expression by LPMCs from patients with CD. These results indicated that T-bet may essentially contribute to Th1 immune responses in CD.
T-bet did not contribute to synergistic augmentation of IFN-γ production by IL-12 and IL-18 stimulation
To investigate which stimuli induce T-bet in CD, we focused on Th1 inducing cytokines such as IL-12, which is secreted by activated macrophages and dendritic cells via activation of innate immune responses. We first measured spontaneous IL-12 production from LPMCs. As previously reported,24 IL-12 production by LPMCs from patients with CD (333.3 (54.7) pg/ml) was significantly (p<0.01) higher than that from patients with UC (48.4 (7.1) pg/ml) or NL (56.7 (11.4) pg/ml) (fig 2A). We next examined whether IL-12 and/or another Th1 inducing cytokine IL-18 could regulate T-bet expression in LPMCs from patients with CD. While IL-12 or IL-18 by themselves increased IFN-γ production by LPMCs from patients with CD, in combination a dramatic effect on IFN-γ production by cells was noted. However, neither single nor combined addition of IL-12/IL-18 significantly upregulated T-bet expression (fig 2C). These results demonstrate that synergistic augmentation of IFN-γ production by IL-12 and IL-18 in CD patients did not require further T-bet upregulation, suggesting a T-bet independent pathway.

T-box expressed in T cells (T-bet) did not contribute to synergistic augmentation of interferon γ (IFN-γ) production by interleukin (IL)-12 and IL-18 stimulation. (A) Lamina propria mononuclear cell (LPMCs) were cultured in medium alone for 48 hours. Production of IL-12 was measured by ELISA in Crohn’s disease (CD) and ulcerative colitis (UC) patients, and in normal controls (NL). **p<0.01. (B) LPMCs from CD patients were stimulated by IL-12 (1 ng/ml) and/or IL-18 (1 ng/ml) for 48 hours. Production of IFN-γ was measured by ELISA (n = 6). (C) T-bet protein expression was assessed by western blotting. Results are representative of three independent experiments.
Induction of T-bet via the T cell receptor (TCR) signalling pathway is necessary for IFN-γ production before IL-12 stimulation
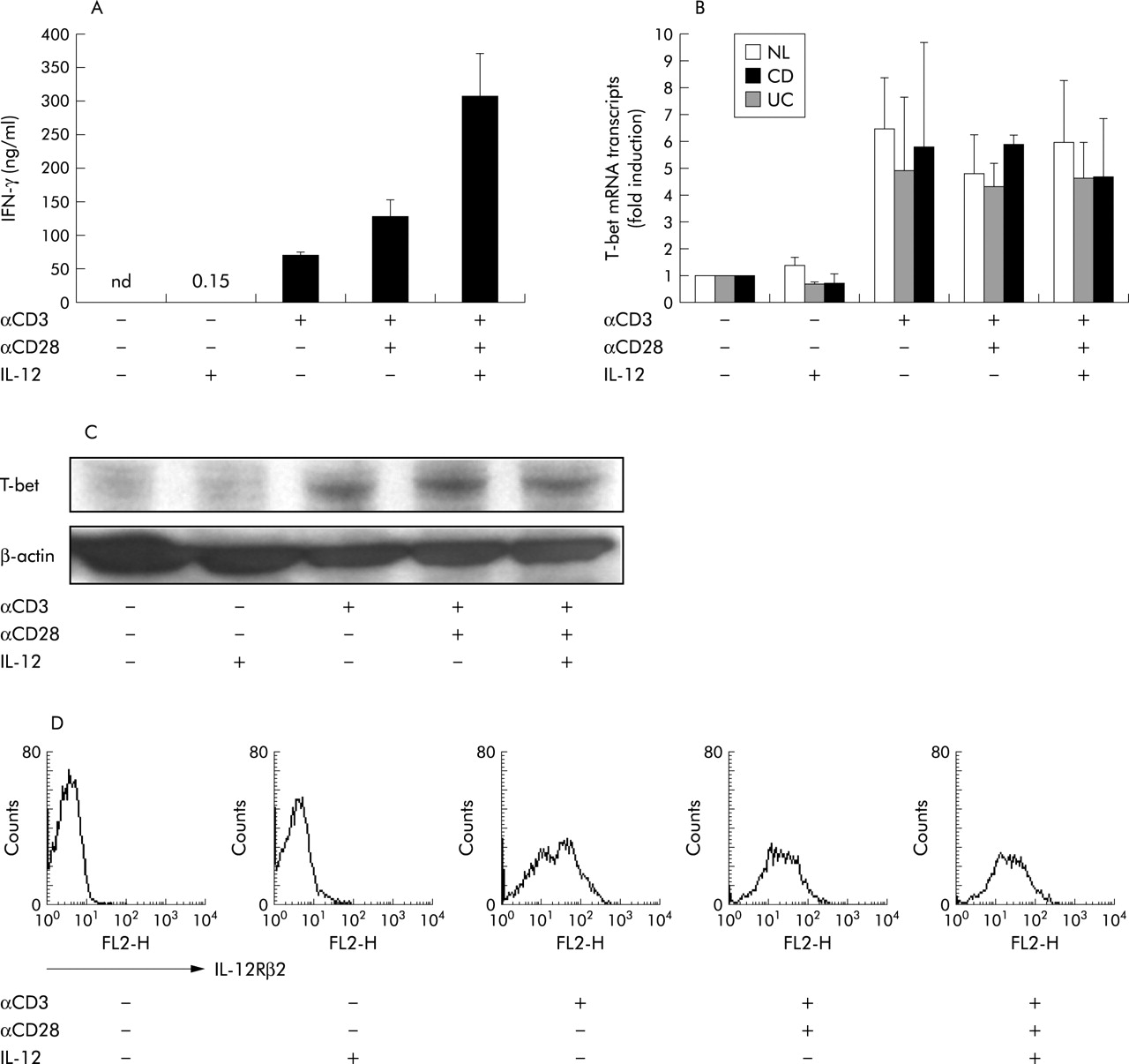
Next, we hypothesised that TCR stimulation was important for T-bet induction early in Th1 mediated immunopathology in CD. Because LPMCs are reported to show activated phenotypes,2 indicating that they have already experienced antigenic stimulation in the intestinal mucosa, we used CD4+ PBMCs in the following experiments. CD4+ PBMCs were stimulated with plate bound anti-CD3 and/or CD28 mAb in the presence or absence of IL-12. After in vitro activation, IFN-γ production and T-bet expression were analysed. As shown in fig 3A, CD4+ PBMCs from CD patients produced little IFN-γ in the presence of IL-12 without plate bound anti-CD3 mAb stimulation. Under these conditions, T-bet was not augmented at either the mRNA or protein level (fig 3B, C). In contrast, anti-CD3 mAb stimulation without exogenous IL-12 induced a large amount of IFN-γ (68.5 (5.0) ng/ml), as well as marked expression of T-bet (fig 3A–C). The level of T-bet induction by anti-CD3 mAb stimulation was not significantly different among CD4+ PBMCs from CD or UC patients, or NL (fig 3B). Consistent with mRNA expression, T-bet protein was augmented by anti-CD3 mAb stimulation in CD4+ PBMCs from patients with CD (fig 3C; similarly in UC and NL, but data not shown). Furthermore, in parallel with T-bet induction by anti-CD3 mAb stimulation, IL-12Rβ2 was also induced in CD4+ PBMCs (fig 3D) and additional stimulation by IL-12 induced a fivefold greater amount of IFN-γ (355.0 (62.2) ng/ml) than anti-CD3 mAb stimulation alone (fig 3A). However, IL-12 costimulation did not induce further T-bet upregulation (fig 3B, C) which was consistent with data regarding LPMCs shown in fig 2C. It is reported that transcription of IL-12Rβ2 was regulated by T-bet in mice.25,26 Collectively, these results indicate that T-bet is induced through TCR stimulation prior to IL-12 signalling for enhanced IFN-γ production; T-bet may not only initiate IFN-γ production but also control the responsiveness to IL-12 through upregulation of IL-12Rβ2.

{kind=link}
{kind=link}
{kind=link}
Induction of T-box expressed in T cells (T-bet) via the T cell receptor (TCR) signalling pathway is necessary for interferon γ (IFN-γ) production before interleukin (IL)-12 stimulation. CD4+ PBMCs were stimulated with anti-CD3 (10 μg/ml)/CD28 (5 μg/ml) and/or IL-12 (10 ng/ml) for 48 hours. (A) Production of IFN-γ was measured by ELISA (n = 3). (B) T-bet mRNA expression was analysed by quantitative real time polymerase chain reaction after stimulation for 12 hours in patients with Crohn’s disease (CD) and ulcerative colitis (UC), and in normal controls (NL). (C) T-bet protein expression was assessed by western blotting. (D) Surface expression of IL-12Rβ2 was analysed by FACS.
DISCUSSION
Although CD represents typical Th1 mediated immune responses, the molecular mechanism involved in differentiation of Th1 cells in CD has not been elucidated. T-bet has been proposed to be the master regulator of Th1 development based on its induction of IFN-γ and repression of Th2 cytokines.19 Furthermore, a study in mice showed that T-bet was STAT-4 independent and acted before IL-12 in Th1 development.25 These observations led us to consider the role of T-bet in human inflammatory bowel diseases, especially in CD. Here we demonstrated that T-bet was expressed strongly in CD4+ LPMCs from CD patients compared with those from UC patients and NL. Neurath et al demonstrated that T-bet was strongly expressed in CD3+ LPMCs in patients with CD while only weak expression was observed in NL and patients with UC.21 A definitive conclusion could not be drawn from their results because CD3+ T lymphocytes included both CD4+ and CD8+ T lymphocytes, and T-bet is critical for IFN-γ production in CD4+ but not in CD8+ T lymphocytes.20 Accordingly, we focused on isolated CD4+ LPMCs to study the role of T-bet in induction of Th1 immune responses in CD. In this study, we showed upregulation of T-bet in CD4+ LPMCs from CD. Increased expression of T-bet in CD was consistent with enhanced production of IFN-γ from LPMCs of CD. Collectively, these results indicate that T-bet may play an essential role in induction of Th1 immune responses in CD.
What regulates T-bet augmentation in CD? We demonstrated that induction of T-bet in human CD4+ PBMCs was mediated by anti-CD3 mAb, but not IL-12, suggesting that antigenic stimulation was essential for T-bet induction. Although some studies have suggested that IFN-γ/STAT-1 signalling is the key pathway for T‐bet expression,26,27 another study revealed that TCR signalling could maintain T-bet expression in committed CD4+ T cells.28 The result indicated that repeated antigenic stimulations were essential for retaining high T-bet expression. It is well known that the gastrointestinal tract is continuously exposed to a large amount of antigens derived from the diet, bacteria, and pathogens. Sustained dysregulated T cell responses to ubiquitous antigens could result in continuous upregulation of T-bet in CD. Hyporesponsiveness (tolerance) against various luminal antigens in normal intestinal mucosa is maintained through anti-inflammatory cytokines such as transforming growth factor-β (TGF-β). It was reported that TGF-β suppressed T-bet expression.21,29 Considering that mucosal T cells from CD patients have been shown to be insensitive to TGF-β inhibition,30 disruption of TGF-β signalling may possibly be involved in overexpression of T-bet as well as hyperreactivity against various luminal antigens. These findings are consistent with the finding that elimination of luminal antigens by elemental diet or decontamination of gut flora can attenuate mucosal inflammation in human CD and murine experimental colitis models.31–34
Although induction of T-bet by anti-CD3 mAb in CD4+ PBMCs was not different between CD and UC, increased expression of T-bet was observed only in CD4+ LPMCs from patients with CD. These data suggest that the cytokine environment in UC is different from that in CD (that is, increased production of IL-5, which is a typical Th2 cytokine).5 It was reported that Th2 skewing conditions suppressed T-bet expression.19 Therefore, in vivo T-bet expression in the presence of chronic gut inflammation may be influenced by the local environment, including cytokine patterns. Further studies would however be necessary to clarify these points.
What then is the role of T-bet in induction of Th1 mediated immunopathology in CD? In the present study, we demonstrated that IL-12 in itself could not induce IFN-γ production from CD4+ PBMCs that did not express T-bet. In contrast, anti-CD3 mAb stimulation induced T-bet expression in CD4+ PBMCs and caused them to produce IFN-γ. Activated CD4+ PBMCs also increased IL-12Rβ2 and responded to IL-12 for further production of IFN-γ. It has been reported that transcription of IL-12Rβ2 is regulated by T-bet in mice.25,26 We and other investigators have previously demonstrated that LPMCs from CD patients expressed high levels of IL-12Rβ2 and responded to IL-12 more strongly than those from NL.35,36 Consistent with previous reports, our data indicate that T-bet is necessary for initiating IFN-γ production, and that it regulates IL-12 responsiveness through IL-12Rβ2 upregulation in CD.
We have demonstrated here that T-bet expression was increased in CD4+ LPMCs from patients with CD. T-bet induced by antigens in the gut lumen initiates IFN-γ production and modulates IL-12 responsiveness through upregulation of IL-12Rβ2. Furthermore, dysregulated production of IL-12/IL-18, as observed specifically in CD patients in our previous and present studies, may intensify Th1 polarisation of the majority of T cells initiated by T-bet. Thus serial actions by antigenic stimulation to induce T-bet in LPMCs and macrophage derived IL-12/18 could be essential for establishing Th1 mediated immunopathology in CD.
Acknowledgments
We thank Professor Daniel Podolsky and Dr Cosmas Giallourakis for critical comments, Drs L Glimcher and S Szabo for providing the antibodies, Professor M Kitajima and Drs M Watanabe, H Hasegawa, Y Iwao, and M Naganuma for providing the specimens, and Dr H Takaishi for helpful discussion. This work was supported in part by grants-in-aid from the Japanese Ministry of Education, Culture, and Science, the Japanese Ministry of Labor, Health, and Welfare, Japan Health Sciences Foundation, Keio University, and Keio University Medical Fund.