Article Text

Abstract
Objective Fibroblast growth factors (Fgfs) are key orchestrators of development, and a role of Fgfs in tissue repair is emerging. Here we studied the consequences of inducible loss of Fgf receptor (Fgfr) 4, the major Fgf receptor (Fgfr) on hepatocytes, alone or in combination with Fgfr1 and Fgfr2, for liver regeneration after PH.
Design We used siRNA delivered via nanoparticles combined with liver-specific gene knockout to study Fgfr function in liver regeneration. Liver or blood samples were analysed using histology, immunohistochemistry, real-time RT-PCR, western blotting and ELISA.
Results siRNA-mediated knockdown of Fgfr4 severely affected liver regeneration due to impairment of hepatocyte proliferation combined with liver necrosis. Mechanistically, the proliferation defect resulted from inhibition of an Fgf15-Fgfr4-Stat3 signalling pathway, which is required for injury-induced expression of the Foxm1 transcription factor and subsequent cell cycle progression, while elevated levels of intrahepatic toxic bile acids were identified as the likely cause of the necrotic damage. Failure of liver mass restoration in Fgfr4 knockdown mice was prevented at least in part by compensatory hypertrophy of hepatocytes. Most importantly, our data revealed partially redundant functions of Fgf receptors in the liver, since knockdown of Fgfr4 in mice lacking Fgfr1 and Fgfr2 in hepatocytes caused liver failure after PH due to severe liver necrosis and a defect in regeneration.
Conclusions These results demonstrate that Fgfr signalling in hepatocytes is essential for liver regeneration and suggest activation of Fgfr signalling as a promising approach for the improvement of the liver's regenerative capacity.
- LIVER REGENERATION
- GROWTH FACTORS
- SIGNALING
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Fibroblast growth factor receptor (Fgfr)1 and Fgfr2 on hepatocytes control compound detoxification in the regenerating liver.
Global loss of Fgfr4 in mice does not cause an obvious defect in liver regeneration after partial hepatectomy.
Loss of Fgf15, the major Fgfr4 ligand, strongly impairs hepatocyte proliferation and causes liver damage after partial hepatectomy.
What are the new findings?
Inducible loss of Fgfr4 in the liver through siRNA-mediated knockdown severely impairs liver regeneration.
Fgf15 levels increase in the serum after partial hepatectomy and most likely activate Fgfr4 on hepatocytes.
Mechanistic studies reveal that an Fgf15-Fgfr4-Stat3-FoxM1 axis controls hepatocyte proliferation in the regenerating liver.
Combined loss of Fgfr1, Fgfr2 and Fgfr4 in hepatocytes causes liver failure after partial hepatectomy, demonstrating that Fgfr signalling is essential for liver regeneration.
How might it impact on clinical practice in the foreseeable future?
Activation of Fgfr4 signalling through treatment with the ligand Fgf15 or low-molecular-weight compounds is a promising approach for the improvement of liver regeneration, in particular in patients with severe liver damage.
Introduction
Fibroblast growth factors (Fgfs) comprise a family of 22 proteins in mammals, which control proliferation, differentiation and survival of various cell types. They exert their functions by activation of four transmembrane tyrosine kinase receptors, designated Fgfr1–Fgfr4.1 ,2 Fgf expression is strongly upregulated after injury to various tissues and organs as initially demonstrated for the skin.3 This is functionally important since inhibition of Fgfr signalling in mice caused impaired wound healing4 and also affected the repair process of other organs.2 ,5 Therefore, Fgfs are promising molecules for the treatment of impaired tissue repair.
The liver is the only organ in adult mammals that can fully regenerate even after severe injury.6 ,7 This remarkable regenerative capacity involves the action of different growth factors, and ligands of the epidermal growth factor receptor as well as hepatocyte growth factor (Hgf) are considered as the major mitogens for hepatocytes in the regenerating liver.7 ,8 Our previous studies also revealed a role of Fgfs in this process since hepatocyte proliferation after two-third (partial) hepatectomy (PH) was impaired in mice expressing a dominant-negative Fgfr2 mutant in hepatocytes.9 Since this mutant inhibits signalling via different Fgfr to a different extent, the role of individual receptors in the regenerative response and their mechanisms of action remained, however, unclear. Therefore, we studied the liver regeneration process in mice lacking Fgfr1 and Fgfr2 in hepatocytes and identified an important function of these receptors in compound detoxification, while the peak in hepatocyte proliferation was only mildly affected.10 By contrast, no obvious regeneration defect after PH was observed in mice with a global Fgfr4 knockout,11 although Fgfr4 is the most abundant Fgfr on hepatocytes with an important function in liver tumorigenesis.12 ,13 Since mice with a global knockout of a gene frequently activate compensatory pathways during prenatal and/or postnatal development, we studied the consequences of an inducible loss of this receptor in adult mice for liver regeneration. We used siRNA formulated into lipid nanoparticles, which allows an efficient knockdown in the liver after a single injection.14 The nanoparticles specifically target hepatocytes due to an apolipoprotein E-dependent mechanism of delivery.15–17 Recently, we demonstrated the usefulness of an siRNA approach to determine the role of β1-integrin in liver regeneration.18 Importantly, the knockdown was only efficient in hepatocytes, but not in non-parenchymal liver cells.18 Using this approach, we determined the consequences of Fgfr4 knockdown for liver regeneration in the presence or absence of Fgfr1 and Fgfr2 in hepatocytes. We demonstrate an important role of Fgfr4 in liver regeneration and an essential role of Fgfr signalling in general for this process.
Materials and methods
Animals
Wild-type mice, mice lacking Fgfr1 and Fgfr2 in hepatocytes10 and mice expressing Cre recombinase in hepatocytes (Alb-Cre mice) were used in this study. They were maintained in a pathogen-free facility under optimal hygiene conditions. Mouse maintenance and all procedures with animals had been approved by the local veterinary authorities of Zurich, Switzerland.
siRNA-mediated knockdown
siRNAs were formulated in lipid nanoparticles (ionisable lipoids), which specifically target hepatocytes due to an apolipoprotein E-dependent mechanism of delivery.15 ,16 ,17 Male mice received formulated siRNA directed against luciferase (Luc) or Fgfr4 (0.5 mg/kg) via tail vein injection in a volume of 5 mL/kg body weight at days 1 and 5. PH was performed at day 10. The following siRNAs were used:
| Fgfr4 | Sense 5′-ccuGAuGcAucGAcAuuuAdTsdT-3′ |
| Antisense 5′-uAAAUGUCGAUGcAUcAGGdTsdT-3′ | |
| Fgfr4-2 | Sense 5′-GGcuGAAAcAcGucGucAudTsdT-3′ |
| Antisense 5′-AUGACGACGUGUUUcAGCCdTsdT-3′ | |
| Luciferase | Sense 5′-cuuAcGcuGAGuAcuucGAdTsdT-3′ |
| Antisense 5′-UCGAAGuACUcAGCGuAAGdTsdT-3′ |
2′-OMe modified nucleotides are in lower case, and phosphorothioate linkages are indicated by ‘s’.
Partial hepatectomy
Eight-week to 10-week-old male mice, which had received food and water ad libitum before surgery, were anaesthetised by inhalation of isoflurane, and PH was performed in the morning between 08:00 and 12:00.10 After PH, mice were injected with buprenorphine for analgesia (Temgesic; Essex Chemie AG, Luzern, Switzerland; 0.1 mg/kg of body weight). They were euthanised by CO2 inhalation, and the remaining liver was removed at different time points after PH. The liver tissue that was removed during PH was considered 0 h time point.
Histology and histomorphometry
Liver samples were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) or in 95% ethanol/1% acetic acid and embedded in paraffin. Sections (3.5 µm) were stained with H&E and photographed. The necrotic area was determined morphometrically. For cell size measurement, acidic ethanol-fixed sections were permeabilised with Triton X-100 and the actin cytoskeleton was stained with rhodamine-coupled phalloidin (Life Technologies, Carlsbad, California, USA). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Sections were mounted with Mowiol.
Identification of proliferating cells in vivo
Proliferating cells were identified by 5-bromo-2′-deoxyuridine (BrdU) labelling 2 h before sacrifice. Liver sections were stained with a BrdU antibody, and labelled cells were counted in four independent microscopic fields (200× magnification) per animal.10
Isolation of parenchymal and non-parenchymal cells from mouse liver
Liver was perfused and digested, and hepatocytes and non-parenchymal cells were separated as described.19
Terminal dUTP nick-end labelling
The In Situ Cell Death Detection Kit (Roche, Basel, Switzerland) was used for the detection of apoptotic cells.
Serum analysis
After sacrifice, blood was taken by heart punctuation. After coagulation, serum was harvested, snap-frozen and analysed using FUJI DRI-CHEM 4000i and the corresponding reagents (Fuji, Tokyo, Japan).
Immunohistochemistry
Paraffin sections were incubated for 30 min in 12% bovine serum albumin in PBS/0.025% NP-40 to block unspecific binding sites. The primary antibody (anti-Ly6G; BD Pharmingen, Allschwil, Switzerland) was incubated overnight at 4°C. Sections were stained using the ABC Vectastain Peroxidase Kit (Vector Laboratories, Burlingame, California, USA) according to the manufacturer's protocol, counterstained with haematoxylin, rehydrated and mounted.
RNA isolation and quantitative RT-PCR analysis
Isolation of total cellular RNA, cDNA synthesis and quantitative RT-PCR (qRT-PCR) were carried out as described previously10 using the following primers:
| Gapdh | Forward: 5′-TCG TGG ATC TGA CGT GCC GCC TG-3′ Reverse: 5′-CAC CAC CCT GTT GCT GTA GCC GTA T-3′ |
| Fgfr4 | Forward: 5′-TTG GCC CTG TTG AGC ATC TTT-3′ Reverse: 3′-GCC CTC TTT GTA CCA GTG ACG-3′ |
| Fgfr1 | Forward: 5′-CAA CCG TGT GAC CAA AGT GG-3′ Reverse: 5′-TCC GAC AGG TCC TTC TCC G-3′ |
| Fgfr2 | Forward: 5′-ATC CCC CTG CGG AGA CA-3′ Reverse: 5′-GAG GAC AGA CGC GTT GTT ATC C-3′ |
| Fgfr3 | Forward: 5′-GTG GCT GGA GCT ACT TCC GA-3′ Reverse: 5′-ATC CTT AGC CCA GAC CGT GG-3′ |
| Foxm1 | Forward: 5′-TGA CAT TGG ACC AAG TGT TTA AGC-3′ Reverse: 5′-TCG TTT CTG CTG TGA TTACCAA GT-3′ |
| Cyclin D1 | Forward: 5′-GCG TAC CCT GAC ACC AAT CTC-3′ Reverse: 5′-ACT TGA AGT AAG ATA CGG AGG GC-3′ |
| Cyclin E1 | Forward: 5′-TCC ACG CAT GCT GAA TTA TC-3′ Reverse: 5′-TTG CAA GAC CCA GAT GAA GA-3′ |
| Cyclin A2 | Forward: 5′-CTT GGC TGC ACC AAC AGT AA-3′ Reverse: 5′-CAA ACT CAG TTC TCC CAA AAA CA-3′ |
| Cyclin B1 | Forward: 5′-GCG TGT GCC TGT GAC AGT TA-3′ Reverse: 5′-CCT AGC GTT TTT GCT TCC CTT-3′ |
| Cyp7a1 | Forward: 5′-GGG ATT GCT GTG GTA GTG AGC-3′ Reverse: 5′-CTG TTG CCC AAC TAA GGT ATG G-3′ |
| Fxr | Forward: 5′-GCTTGATGTGCTACAAAAGCTG-3′ Reverse: 5′-CGTGGTGATGGTTGAATGTCC-5′ |
| Tbp | Forward: 5′-AGA ACA ATC CAG ACT AGC AGC A-3′ Reverse: 5′-GCC CTC TTT GTA CCA GTG ACG-3′ |
Cell culture
Mouse immortalised hepatocytes (AML12 cells), Hepa1-6 hepatoma cells and C2C12 myoblasts were cultured in DMEM/10% FCS/penicillin/streptomycin.
Preparation of protein lysates and western blot analysis
Frozen tissue was homogenised in T-PER tissue protein extraction reagent (Pierce, Rockford, Illinois, USA) containing Complete Protease Inhibitor Cocktail and PhosSTOP Phosphatase Inhibitor Cocktail (Roche). Lysates were cleared by sonication and centrifugation.
Cultured cells were lysed in 1× Laemmli buffer at 95°C. Proteins were analysed by western blotting using antibodies against Fgfr4 (R&D Systems, Minneapolis, Minnesota, USA), Gapdh (HyTest, Turku, Finland), Foxm1 and Cyp7a1 (Santa Cruz, Santa Cruz, California, USA), phospho-STAT3 (Tyr-705) and total-STAT3 (both from Cell Signaling, Beverly, Massachusetts, USA).
Cell cycle analysis of cultured cells
Incorporation of 5-ethynyl-2′-deoxyuridine (EdU) into DNA was monitored by flow cytometry using the Click-iT EdU Flow Cytometry Assay kit (Invitrogen, Basel, Switzerland). The same staining provided information on the cell cycle state of the cells. Hepa1-6 cells were cultured in 6-well plates and transfected with Luc or Fgfr4 siRNA (10 nM siRNA) using lipofectamine (Sigma, Buchs, Switzerland). After 48 h incubation, EdU was added to a final concentration of 10 μM. Four hours later, cells were removed by 5 min accutase treatment (1:10, Lucerna-Chem, Luzern, Switzerland). Cells were analysed using a BD LSRII Fortessa (BD, Allschwil, Switzerland) and FlowJo V.8.7 software (Treestar, Ashland, Oregon, USA).
Determination of Fgf15 serum levels
Serum levels of Fgf15 were determined by ELISA (Antibodies-online.com, Aachen, Germany) according to the manufacturer`s instructions.
Cytokine multiplex ELISA
Cytokine concentrations were measured in 50 μg liver lysate using the magnetic bead multiplex suspension array system (Bio-Plex ProT Mouse Cytokine 23-plex Assay; BioRad; Hercules, California, USA). Data were analysed using the MagPix instrument and Bioplex Software (BioRad).
Bile acid quantification
Bile acids were quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS).20 Instead of serum, liver homogenates corresponding to 1.25 mg wet weight were analysed.
Statistical analysis
Statistical analysis was performed using the Prism6 software (GraphPad Software, San Diego, California, USA). Quantitative data are expressed as mean±SEM. Significance was calculated using Mann–Whitney U test; *p≤0.05, **p≤0.01, ***p≤0.001.
Results
Impaired liver regeneration after siRNA-mediated knockdown of Fgfr4
We used lipid-based nanoparticles to deliver Fgfr4 siRNA to hepatocytes. Forty-four Fgfr4 siRNAs were tested for their in vitro efficacy using C2C12 mouse myoblasts, which express high levels of Fgfr4. The most potent siRNAs—Fgfr4 and Fgfr4-2 (see online supplementary figure S1A)—were packaged into nanoparticles. siRNA against Luc was used as control. Nanoparticles were injected into the tail vein at day 1 and day 5, and mice were subjected to PH 5 days later (figure 1A). The initial analysis of non-injured liver and liver 48 h after PH was performed with wild-type and Alb-Cre mice, and the results were identical (data not shown). Since it was the ultimate goal to knock down Fgfr4 in mice lacking Fgfr1 and Fgfr2 in hepatocytes (Alb-R1/R2 mice10), Alb-Cre mice were used for all follow-up studies since they are the appropriate control for Alb-R1/R2 mice.

Impaired liver regeneration in mice after knockdown of Fgfr4 in hepatocytes. (A) Schematic representation of the experimental setup. Mice were injected twice with Fgfr4 or Luc siRNA with a time interval of 5 days. After additional 5 days, they were subjected to two-third (partial) hepatectomy (PH). (B) Knockdown of Fgfr4 was verified by qRT-PCR using RNA from total liver or cultured primary hepatocytes. N=3–7 per treatment group. Expression levels in mice treated with Luc siRNA were arbitrarily set as 1. (C) Activities of ALT and AST in the serum were determined in non-injured mice (n.i.) and at different time points after PH (N=3–9). (D) Morphometric analysis of the necrotic damage using H&E-stained liver sections at different time points after PH in mice injected with Fgfr4 siRNA and at 48 h in mice injected with Fgfr4-2 siRNA. N=4–9. Representative sections from mice 48 h after PH are shown below. Necrotic areas are encircled with dotted lines. Scale bar: 100 μM. (E) BrdU-positive cells were counted in liver sections at different time points after PH in mice injected with Fgfr4 siRNA and at 48 h in mice injected with Fgfr4-2 siRNA. N=4–9. Representative sections stained with a BrdU antibody are shown for the 48 h time point. Scale bar: 50 μM. (F) Levels of Fgf15 in the serum were determined in non-injected mice and at different time points after PH. N=3–5. All bars represent mean ±SEM. ALT, alanine transaminase; AST, aspartate transaminase; BrdU, 5-bromo-2′-deoxyuridine; Fgfr, fibroblast growth factor receptor; Luc, luciferase.
Efficient knockdown of Fgfr4 was verified by qRT-PCR using RNAs from total liver or cultured primary hepatocytes and by western blotting using total liver lysates (figure 1B). The similar knockdown efficiency seen with lysates from total liver and from isolated hepatocytes is consistent with the strong expression of Fgfr4 by hepatocytes, but not by non-parenchymal cells (see online supplementary figure S1B). Efficient knockdown by Fgfr4 siRNA was confirmed for all time points and by Fgfr4-2 siRNA for the 48 h time point, while expression of other Fgfr was not affected (see online supplementary figure S1C–E). Knockdown of Fgfr4 did not cause liver injury within the analysed time frame as revealed by normal serum levels of aspartate and alanine aminotransferases (AST and ALT) (figure 1C) and the normal liver histology (see online supplementary figure S1F). Most control mice survived the first week after PH, whereas 25% of the mice with Fgfr4 knockdown died between 48 and 216 h after surgery (see online supplementary figure S2A). ALT and AST levels were increased compared with control mice at 24 and 48 h after PH (figure 1C). Severe necrosis was observed up to 48 h, in particular around portal areas (figure 1D), and there was enhanced steatosis, neutrophil infiltration and an increase in the number of apoptotic cells (see online supplementary figure S2B–D). Their small nuclei suggest that the apoptotic cells are predominantly non-parenchymal cells. The damage was, however, repaired during the following days (figure 1D). The peak in hepatocyte DNA replication that occurred in control mice at 48 h after PH was not observed after Fgfr4 knockdown. There was no compensatory hyperproliferation at later time points (figure 1E) and also no increase compared with control at 36 h (data not shown). Since we still observed a strong knockdown after 9 days (see online supplementary figure S1C), it is unlikely that cells with inefficient knockdown preferentially contributed to regeneration.
Similar regeneration defects were observed with Fgfr4-2 siRNA (figure 1D, E), and the more efficient siRNA (Fgfr4) (see online supplementary figure S1A) caused a stronger phenotype compared with the less efficient siRNA (Fgfr4-2). Mice injected with Luc siRNA did not exhibit necrosis (figure 1D), and the time course of regeneration as well as AST/ALT levels after PH were similar as in mice not injected with any siRNA18 (and data not shown). This finding demonstrates lack of toxicity of the nanoparticles even after liver injury.
Levels of the major Fgfr4 ligand, Fgf15,21 strongly increased in the serum of control mice within 6–24 h after PH, most likely as a result of release of intrahepatic bile acids into the intestine after surgery. This increase was much more pronounced after Fgfr4 knockdown (figure 1F), possibly due to higher levels of bile acids that stimulate Fgf15 expression in the intestine,21 since activation of Fgfr4 by intestinal Fgf15 suppresses bile acid production.11 Indeed, expression of cholesterol 7 α-hydroxylase (Cyp7a1), the rate-limiting enzyme in bile acid production,22 was increased in mice with Fgfr4 knockdown prior to PH (see online supplementary figure S3A), and the concentrations of intrahepatic bile acids, in particular of tauro-conjugated bile acids, were significantly higher than in control mice at 24 and 48 h after PH (table 1 and see online supplementary figure S3B). The increase in bile acids in the liver during regeneration provides an explanation for the liver necrosis since bile acids can damage cell membranes due to their detergent properties.23 The necrosis is most likely not due to increased fragility of bile ductules in the absence of Fgfr4 since staining with a pan-keratin antibody revealed no obvious defect in the integrity of bile ducts at periportal locations (see online supplementary figure S3C).
Enhanced levels of bile acids in mice with Fgfr4 knockdown
Loss of Fgfr4 induces compensatory hepatocyte growth
In spite of the defect in DNA replication by hepatocytes, the liver to body weight ratio was only mildly reduced in the Fgfr4 knockdown mice 96 and 216 h after PH (figure 2A), suggesting that cellular hypertrophy compensates at least in part for the proliferation defect. Indeed, hepatocytes of these mice were significantly larger at 216 h after PH compared with control mice, and more cells were binuclear at this time point (figure 2B, C). There was also a strong reduction in mitotic figures 48 h after PH. However, this was followed by an increase at 72 h (figure 2D), demonstrating that cell division eventually occurred, although with a severe delay.

Compensatory growth of hepatocytes after PH in mice with Fgfr4 knockdown. (A) Liver to body weight ratio in non-injured mice and at different time points after PH in mice injected with Fgfr4 or Luc siRNA. N=3–9 mice per treatment group. (B) Size of hepatocytes as determined on sections stained with rhodamine-coupled phalloidin. (C) The percentage of binucleated cells was determined on sections stained with rhodamine-coupled phalloidin. (D) Cells with mitotic figures (indicated by arrows in the representative stainings) were quantified in H&E-stained sections. Approximately 50 cells from three to seven mice were counted in (B–D). Magnification bar: 50 μm. Bars represent mean±SEM. Fgfr, fibroblast growth factor receptor; Luc, luciferase; PH, partial hepatectomy.
Fgfr4 signalling is important for Stat3 activation and PH-induced expression of the forkhead box protein M1 (Foxm1)
Consistent with the defect in cell cycle progression upon Fgfr4 knockdown, expression of cyclins A2 and B1 was reduced at 48 h after PH compared with Luc siRNA injected mice, while cyclins D1 and E1 were only moderately affected (figure 3A–D). Upregulation of Foxm1, a transcription factor with a crucial role in hepatocyte DNA replication and mitosis after PH,24 occurred 48 h after PH in control mice, but to a much lesser extent in Fgfr4 knockdown mice (figure 3E). This was not due to alterations in the expression of the farnesoid X receptor (Fxr), a direct activator of the Foxm1 gene (figure 3F). However, levels of phosphorylated (activated) Stat3, a positive regulator of the Foxm1 gene,25 were much lower in Fgfr4 knockdown mice at 24 h after PH compared with controls, while the early Stat3 activation was not affected (figure 3G). This was not a consequence of interleukin (IL)-6 deficiency since serum levels of IL-6 and its upstream regulator tumour necrosis factor-α were even increased in mice with Fgfr4 knockdown at this time point (see online supplementary figure S4A,B). Rather, it seems to be a direct consequence of the reduction in Fgfr4 levels since Fgfr4 knockdown in mouse hepatoma cells also reduced Stat3 phosphorylation. Concomitantly, DNA synthesis was reduced and cells in G1 accumulated, whereas those in S or G2 phase were reduced (see online supplementary figure S5A–C). Furthermore, levels of Foxm1 and cyclin A2 and B1 mRNAs were strongly reduced in Hepa1-6 and AML12 cells upon transfection with Fgfr4 and Fgfr4_2 siRNAs (see online supplementary figure S5D and data not shown).

Cell cycle progression is impaired in mice with Fgfr4 knockdown. (A–F) qRT-PCR analysis of different cyclins and of Foxm1 and Fxr using RNAs from liver at different time points after PH. N=3–9 per time point and treatment group. Expression levels in non-injured liver of mice injected with Luc siRNA were arbitrarily set as 1. (E and G) Total liver lysates of mice prior to and at different time points after PH were analysed by western blotting for expression of Foxm1 and Gapdh (E), or total and phosphorylated Stat3 (G). The ratio between phosphorylated and total Stat3 is shown. All bars represent mean±SEM. Fgfr, fibroblast growth factor receptor; Luc, luciferase; PH, partial hepatectomy.
Fgfr signalling is essential for liver regeneration
Finally, we determined whether Fgfr1 and Fgfr2 prevent a more severe phenotype in Fgfr4 knockdown mice. For this purpose, we injected Luc or Fgfr4 siRNA into Alb-R1/R2 mice or Alb-Cre mice. Alb-R1/R2 mice showed impaired expression of genes required for compound detoxification, resulting in liver necrosis and an almost 50% reduction in survival upon PH when ketamine/xylazine was used for anaesthesia.10 Since the extent of liver injury and the resulting mortality were much lower with isoflurane anaesthesia,10 we used these conditions for our experiments. While Alb-R1/R2 mice with Fgfr4 knockdown (Alb-R1/R2-Fgfr4 mice) survived the first two days after PH, they subsequently showed signs of liver failure and they died or had to be sacrificed due to health problems (figure 4A). BrdU staining and expression analysis of cyclins A2 and B1 revealed strongly reduced hepatocyte proliferation in the remaining mice at 72 h, and at this time we also observed massive necrosis, liver damage and steatosis (figure 4B–E). In addition, Fgf15 serum levels were dramatically increased (figure 4E), indicating high levels of bile acids that reach the intestine. Indeed, levels of intrahepatic bile acids were already higher prior to PH in Alb-R1/R2-Fgfr4 mice and dramatically increased at 72 h after PH when liver failure occurred (figure 4E and see online supplementary table S1). Thus, in contrast to mice deficient in only Fgfr4, levels of intrahepatic bile acids did not decrease after 48 h in Alb-R1/R2-Fgfr4 mice, and the animals were not able to repair the necrotic damage. These results demonstrate that Fgfr signalling in hepatocytes is essential for liver regeneration.
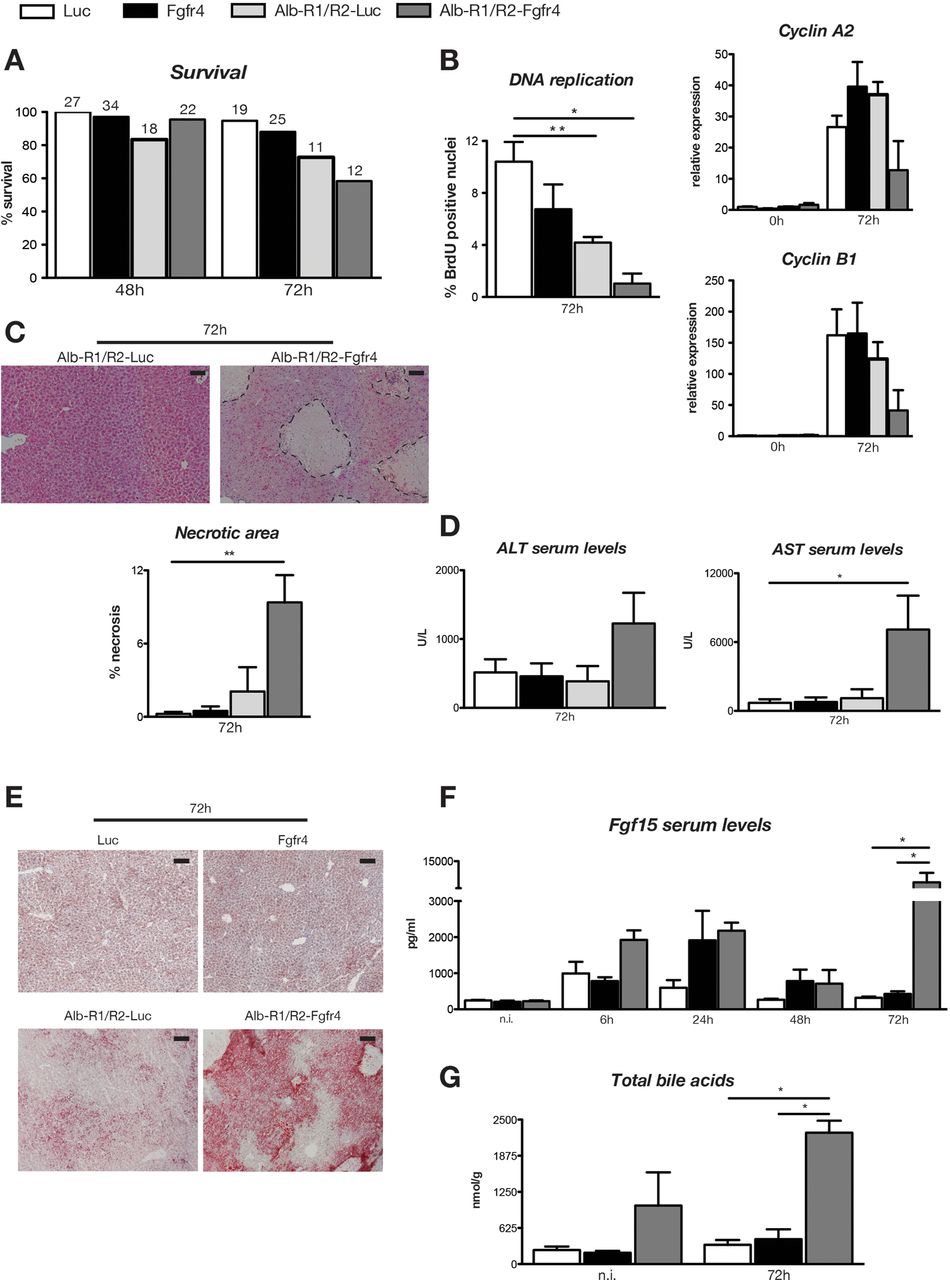
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fgfr signalling is essential for liver regeneration. Alb-R1/R2 mice or Alb-Cre control mice were injected with Fgfr4 or Luc siRNA and subjected to PH. Survival of mice of all genotypes and treatment groups is shown on (A). The number on top of the bars indicates the number of mice included in the analysis at each time point. (B) Left panel: BrdU-positive cells were counted in liver sections 72 h after PH in Alb-R1/R2 mice and Alb-Cre control mice injected with Fgfr4 or Luc siRNA. N=4–9 per time point and treatment group. Right panels: qRT-PCR analysis of cyclins A2 and B1 using RNAs from liver at 0 h and 72 h after PH. N=3–9 per time point and treatment group. Expression levels in mice injected with Luc siRNA 0 h after PH were arbitrarily set as 1. N=4–9. (C) The necrotic area (encircled with dotted line) was determined in H/E-stained sections. N=4–9. (D) AST and ALT activities in the serum of mice of all treatment groups at 72 h after PH. N=4–9. (E) Hepatic steatosis as determined by Oil Red O staining of liver sections. Representative sections from 3–10 mice per treatment group are shown. (F) Serum levels of Fgf15 at different time points after PH. N=3–5. (G) Levels of total intrahepatic bile acids were determined using liquid chromatography—mass spectrometry. N=3–5. All bars represent mean±SEM. ALT, alanine transaminase; AST, aspartate transaminase; BrdU, 5-bromo-2′-deoxyuridine; Fgfr, fibroblast growth factor receptor; Luc, luciferase; PH, partial hepatectomy.
Discussion
We demonstrate a severe impairment of liver regeneration upon knockdown of Fgfr4 in hepatocytes, consistent with the important role of Fgfr4 in liver tumorigenesis through its effect on hepatoma cell proliferation and survival.12 Thus, targeting this receptor is a potential treatment option for this malignancy.13 ,26 Surprisingly, however, mice with a global Fgfr4 knockout exhibited no defect in hepatocyte proliferation after PH, and recovery of their liver mass was not affected.11 The different results obtained with Fgfr4 knockout versus knockdown mice may result from activation of compensatory mechanisms in the knockout mice during prenatal or postnatal development. They are unlikely caused by off-target effects of the siRNA since the same abnormalities were observed with two different Fgfr4 siRNAs. In addition, toxicity of the nanoparticles was excluded since we did not observe liver abnormalities in mice after injection of nanoparticles with Luc siRNA—not even after PH. Most importantly, loss of Fgf15 caused similar alterations in liver regeneration as seen after Fgfr4 knockdown,27 ,28 and loss of Fxr in the intestine also resulted in impaired hepatocyte proliferation after PH, which was rescued by adenoviral overexpression of Fgf15.29 However, the strong lethality observed after PH in Fgf15 knockout mice27 was not observed in Fgfr4 knockdown animals and we also did not find a reduction in Fxr or cyclin D1 expression after Fgfr4 knockdown. Fgf15 can also activate the IIIc variants of Fgfr1, Fgfr2 or Fgfr3,30 which are expressed on non-parenchymal cells, and this may contribute to the effect of Fgf15 on liver regeneration. Hepatocytes only express the IIIb variants of Fgfr1 and Fgfr2,10 which are most likely not activated by Fgf15. However, we cannot fully exclude the possibility that Fgf15 activates these receptors in hepatocytes in vivo. Finally, activation of Fgfr4 by Fgf15 in other organs, which are not targeted by the siRNA, may contribute to the effect of Fgf15 on liver regeneration. However, it seems most likely that very low levels of Fgfr4 that remain in the liver of the siRNA-treated mice are sufficient to maintain normal expression levels of cyclin D1 and Fxr and to allow survival. Alternatively, differences in the analgesia or anaesthesia, the surgical procedure and/or the genetic background may contribute to the differences in liver regeneration between Fgfr4 knockdown and Fgf15 knockout mice.
Our mechanistic studies combined with published data strongly suggest that an Fgf15-Fgfr4-Stat3-Foxm1 axis controls hepatocyte DNA replication and proliferation in the regenerating liver. This hypothesis is based on the following findings: (i) Fgf15 serum levels strongly increased after PH, (ii) loss of Fgf15 caused a similar regeneration defect as knockdown of Fgfr4, (iii) activation of Stat3 and induction of Foxm1 expression by PH were strongly reduced upon knockdown of Fgfr4 in the liver and (iv) knockdown of Fgfr4 in cultured hepatoma cells also reduced the levels of pStat3 and concomitantly resulted in accumulation of cells in G1 and in reduction of DNA synthesis. We propose that the failure to induce Foxm1 expression after PH is primarily responsible for the impaired DNA replication in hepatocytes since Foxm1-deficient mice showed similar abnormalities and also lacked the strong peak of hepatocyte DNA replication that is normally seen 36–48 h post PH.24 By contrast, these mice did not show liver necrosis, indicating that the necrosis seen after PH in Fgfr4 knockdown mice is Foxm1 independent. In addition, it is obviously not due to major damage of the portal bile ductules. Rather, it seems most likely that the effect of Fgfr4 on bile acid metabolism11 ,31 underlies this phenotype. Similar as in Fgf15-deficient mice, this leads to accumulation of cytotoxic bile acids in the liver after PH. The enhanced expression of Cyp7a1 in the non-injured liver of control mice compared with Fgfr4 knockdown mice provides a partial explanation for this finding. However, the PH-induced down-regulation of Cyp7a1 expression32 was not affected in Fgfr4 knockdown mice. This down-regulation was shown to depend on Hgf,33 which was normally expressed in Fgfr4 knockdown mice (see online supplementary figure S6A). Therefore, additional mechanisms, such as defects in the PH-induced regulation of different transporters,23 ,32 are likely to contribute to the accumulation of bile acids in the liver, which remain to be determined. Consistent with the increase in bile acids, which stimulate expression of Fgf15 in the intestine via activation of Fxr,21 serum levels of Fgf15 were much higher in Fgfr4 knockdown mice compared with Luc siRNA-injected mice after PH.
The most dramatic result of our study was the liver failure that occurred between 48 and 72 h after PH in Alb-R1/R2-Fgfr4 mice. This resulted from severe liver necrosis, which—in contrast to mice with loss of only Fgfr4 or Fgfr1/Fgfr2—could not be repaired. The necrosis is most likely the consequence of the prolonged increase in intrahepatic bile acids after PH, which causes continuous cell damage. The molecular mechanisms underlying this strong increase are as yet unknown since Cyp7a1 expression was not significantly altered in Alb-R1/R2-Fgfr4 mice prior and following PH (see online supplementary figure S6B), possibly due to activation of compensatory mechanisms. Therefore, Fgfr deficiency seems to affect other steps in bile acid metabolism and/or transport, which should be addressed in future studies. The bile acid toxicity in combination with the repair defect ultimately resulted in liver failure. The severe lipid accumulation that we observed after PH is likely to further contribute to the severe regeneration phenotype since hepatic steatosis has been shown to affect hepatocyte proliferation after PH.34 ,35
Taken together, our results revealed that Fgfs and their receptors are essential orchestrators of liver regeneration. This is likely to be of major medical relevance because the remarkable capacity of the liver to regenerate is insufficient after chronic injury or upon removal of particularly large amounts of liver tissue. In the latter case, the remaining liver tissue is too small to fulfil the important functions of this organ in metabolism and detoxification, ultimately resulting in small-for-size syndrome and liver failure. Remarkably, the reduced survival that occurs after extended PH (86%) was partially rescued by adenoviral delivery of Fgf15.27 Furthermore, inducible overexpression of Fgf7 in transgenic mice prevented chronic liver damage and even stimulated repair after chronic injury, most likely through the activation of Fgfr2-IIIb on liver progenitor cells.36 Thus, Fgfs are emerging as promising therapeutics for the prevention and/or treatment of liver damage and for the improvement of the regeneration process in this organ.
Acknowledgments
We thank Dr Lucia Bautista Borrego, Dr Michael Meyer, Dr Michèle Telorack, Beat Siegenthaler, Lucia Pelloni and Christiane Born-Berclaz (ETH Zurich) for experimental help, Dr David Ornitz (Washington University St. Louis, USA) for providing conditional Fgfr1/Fgfr2 knockout mice and for helpful suggestions, Dr Achim Weber (University of Zurich) for support, and Sol Taguinod (ETH Zurich) for help with animal care. This work was supported by the Swiss National Science Foundation (310030_132884 to SW), a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft (to TS), and a predoctoral fellowship from the Janggen-Pöhn Stiftung (to MB).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Contributors SP-A, MB, RLB, LP, TR, FB, GL and TS performed the experiments and analysed the data. CH was involved in the bile acid analysis, VK coordinated the siRNA research, SW wrote the manuscript and designed the project together with SP-A. DMO provided conditional Fgfr1/Fgfr2 knockout mice. Achim Weber supported the work of Friederike Böhm. Lucia Bautista Borrego, Michael Meyer, Michele Telorack, Lucia Pelloni and Beat Siegenthaler helped with some of the experiments.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.