Article Text

Abstract
Objective The bacterial intestinal microbiota plays major roles in human physiology and IBDs. Although some data suggest a role of the fungal microbiota in IBD pathogenesis, the available data are scarce. The aim of our study was to characterise the faecal fungal microbiota in patients with IBD.
Design Bacterial and fungal composition of the faecal microbiota of 235 patients with IBD and 38 healthy subjects (HS) was determined using 16S and ITS2 sequencing, respectively. The obtained sequences were analysed using the Qiime pipeline to assess composition and diversity. Bacterial and fungal taxa associated with clinical parameters were identified using multivariate association with linear models. Correlation between bacterial and fungal microbiota was investigated using Spearman's test and distance correlation.
Results We observed that fungal microbiota is skewed in IBD, with an increased Basidiomycota/Ascomycota ratio, a decreased proportion of Saccharomyces cerevisiae and an increased proportion of Candida albicans compared with HS. We also identified disease-specific alterations in diversity, indicating that a Crohn's disease-specific gut environment may favour fungi at the expense of bacteria. The concomitant analysis of bacterial and fungal microbiota showed a dense and homogenous correlation network in HS but a dramatically unbalanced network in IBD, suggesting the existence of disease-specific inter-kingdom alterations.
Conclusions Besides bacterial dysbiosis, our study identifies a distinct fungal microbiota dysbiosis in IBD characterised by alterations in biodiversity and composition. Moreover, we unravel here disease-specific inter-kingdom network alterations in IBD, suggesting that, beyond bacteria, fungi might also play a role in IBD pathogenesis.
- INFLAMMATORY BOWEL DISEASE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Video abstract
Significance of this study
What is already known on this subject?
The bacterial intestinal microbiota is unbalanced in IBDs and plays a role in its pathogenesis.
The fungal microbiota has been poorly studied despite several clues of its role in IBD pathogenesis.
Card9 and Dectin1, two key molecules involved in the innate immunity against fungi, strongly influence mice susceptibility to intestinal inflammation and the fungal microbiota.
What are the new findings?
The faecal fungal microbiota is imbalanced in patients with IBD.
The concomitant analysis of the bacterial microbiota in the same subjects showed many correlations between bacterial and fungal components with differences between IBD and healthy subjects, suggesting the existence of disease-specific inter-kingdom alterations.
How might it impact on clinical practice in the foreseeable future?
These results support the role of fungal microbiota in IBD pathogenesis and indicate a new potential therapeutic target.
Introduction
Crohn's disease (CD) and UC, the two primary types of IBD, are lifelong conditions that usually affect young subjects and substantially alter their quality of life. The exact pathogenesis of IBD remains unknown; however, studies over the last decade have demonstrated that IBD involves an altered immune response towards gut microbiota in genetically predisposed subjects and under the influence of environmental factors. The bacterial microbiota in IBD has been thoroughly investigated, and several groups worldwide observed a bacterial dysbiosis (an imbalance in composition) that is characterised by a reduced biodiversity, a decrease in some bacteria belonging to the Firmicutes phylum (such as Faecalibacterium prausnitzii) and an increase in bacteria belonging to the Proteobacteria phylum such as Escherichia coli.1–5 However, other microorganism types colonising the human gut have not been thoroughly investigated. With the exception of a recent study highlighting the possible role of the enteric virome in IBD,6 the data are scarce, particularly regarding fungal microbiota. Fungi have long been suspected in IBD pathogenesis. Many years ago, antibodies directed against Saccharomyces cerevisiae mannan (Anti Saccharomyces cerevisiae antibody (ASCA)) were shown to be associated with CD. Moreover, several IBD-associated genes, such as Card9, are involved in immune responses to fungi.7 In mice, gut inflammation promotes fungi proliferation;8 conversely, some fungi can modulate susceptibility to inflammation in a negative (Candida albicans) or positive (Saccharomyces boulardii) manner.8–11 Finally, mice lacking major genes involved in fungi sensing, such as Dectin-1 or Card9, have an increased fungal microbiota load and are more susceptible to colitis.12 ,13 These data suggest a link between fungal microbiota and IBD pathogenesis.
Here, we characterised the fungal microbiota in both healthy subjects (HS) and patients with well-phenotyped IBD using high-throughput sequencing technology. In the corresponding patients, we also determined the bacterial microbiota composition and the sequence of 22 single-nucleotide polymorphisms (SNPs) in genes known to be involved in fungal susceptibility. We observed a clear fungal dysbiosis in patients with IBD. Moreover, a correlation analysis suggested altered inter-kingdom relations in IBD. Finally, while somewhat lacking in power, our genotype–fungal microbiota analysis suggested that genes may be a driving factor of the fungal microbiota dysbiosis in IBD.
Overall, the data presented in this study represent the most comprehensive analysis of fungal microbiota in patients with IBD to date and provide a rationale to support the role of fungal microbiota in IBD pathogenesis. These data thus pave the way for intervention studies targeting fungal microbiota.
Results
Bacterial dysbiosis in IBD
Our study population was composed of 235 patients with well-phenotyped IBD and 38 HS (see online supplementary 1). We first analysed the bacterial fraction of the microbiota using 16S sequencing. A beta diversity analysis showed a clustering of samples according to disease phenotypes (figure 1A, B, online supplementary figure S1). Compared with HS samples, the alpha diversity (assessed using four different indexes) was significantly decreased in UC and CD and particularly in samples from patients in flare (figure 1C, D, online supplementary figure S2). In all phenotypes, the bacterial microbiota was dominated by bacteria from Firmicutes, Bacteroidetes and Proteobacteria phyla (figure1E, F, online supplementary figure S3). These data are in accordance with the published literature and validate the quality of our cohort and the methods used.
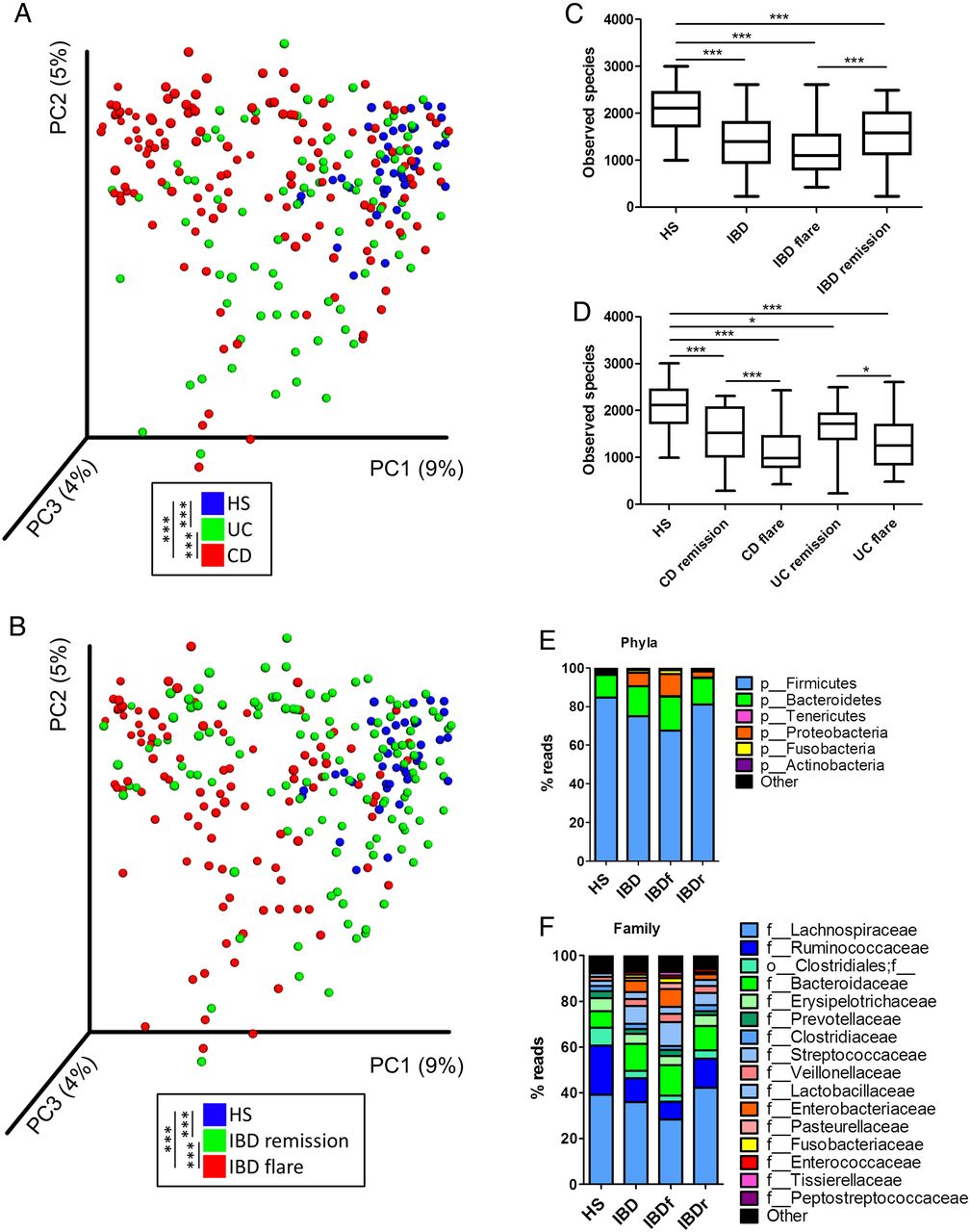
Altered bacterial microbiota biodiversity and composition in IBD. (A and B) Beta diversity. Principal coordinate analysis of Bray–Curtis distance with each sample coloured according to the disease phenotype. PC1, PC2 and PC3 represent the top three principal coordinates that captured most of the diversity. The fraction of diversity captured by the coordinate is given as a percentage. Groups were compared using Permanova method. (C and D) Observed species number describing the alpha diversity of the bacterial microbiota in the various groups studied (Kruskal–Wallis test with Dunn's multiple comparison test). (E and F) Global composition of bacterial microbiota at the phyla and family levels. Healthy subjects (HS) and patient subgroups are labelled on the x-axis and expressed as the relative operational taxonomic unit (OTUs) abundance for each group. In all panels: *p<0.05; **p<0.01; ***p<0.001. CD, Crohn's disease.
Altered fungal microbiota diversity in IBD
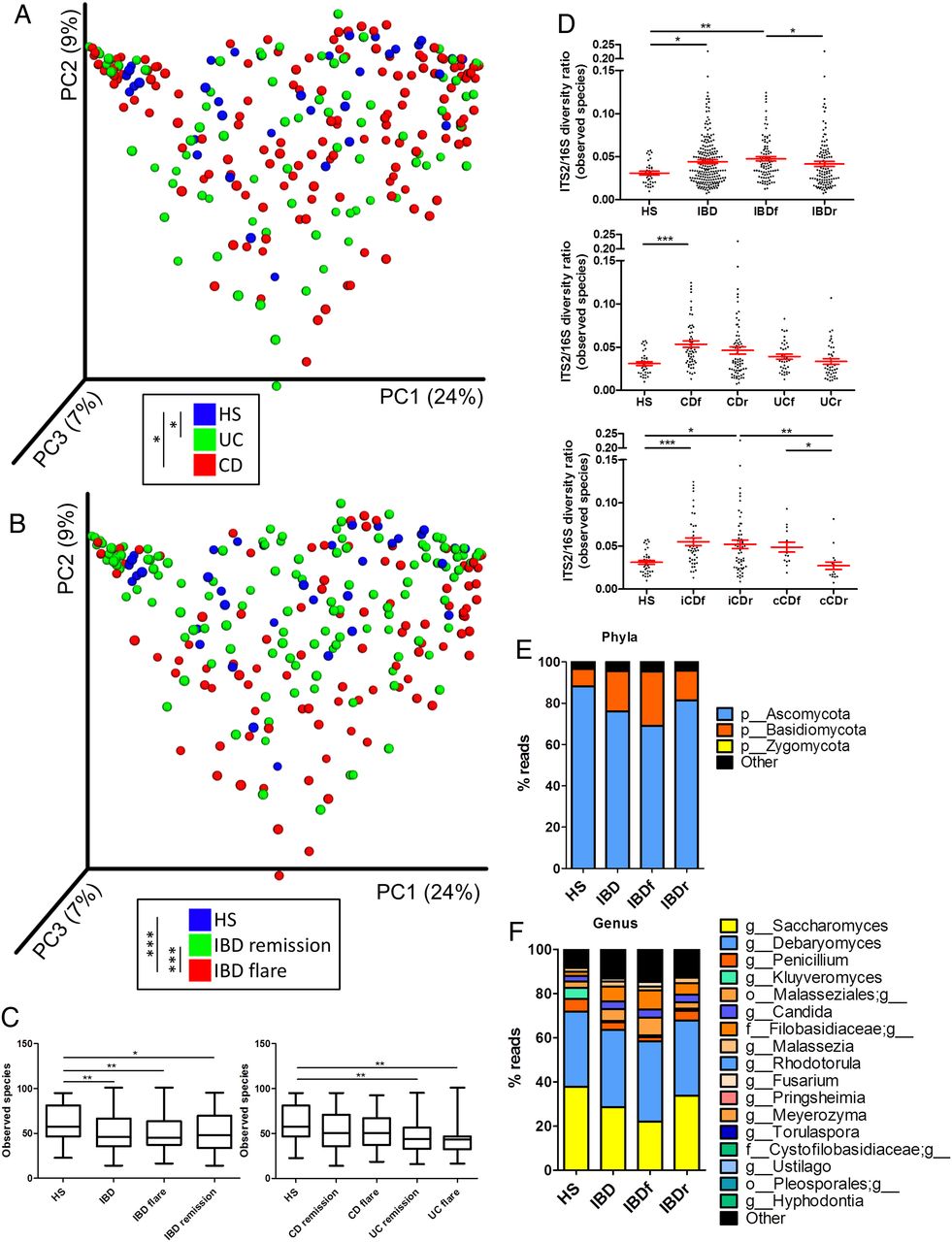
Using ITS2 sequencing, we then assessed the composition of the fungal microbiota in our population. The clustering between the samples according to disease phenotype was weaker than with bacterial microbiota (figure 2A, B, online supplementary figure S4A). Notably, no statistically significant difference was observed between samples from patients with CD and UC or between samples from patients with IBD in remission and HS. However, a difference was observed between samples from patients with IBD in flare and HS (p=0.0008) or patients with IBD in remission (p=0.0007). This fungal microbiota-specific signal in flare was observed with a higher intensity in UC (p=0.0002) than in CD (p=0.006; see online supplementary figure S4B-C). Similar to the results found with the bacterial microbiota, the alpha diversity of the fungal microbiota was decreased in patients with IBD (figure 2C, online supplementary figure S5A–D). This feature was primarily found in samples from patients with UC, whereas fungi diversity was largely similar among the HS and patients with CD. To explore the equilibrium between bacteria and fungi diversity in the gut, we then determined the fungi-to-bacteria diversity ratio. This ratio was increased in IBD samples and particularly in CD and flares (figure 2D, online supplementary figure S5E–G). The highest ratio was found among patients with CD and demonstrated ileal involvement. In both HS and patients with IBD, the fungal microbiota was dominated by fungi from the Ascomycota and Basidiomycota phyla with some variations according to disease phenotype (figure 2E, F, online supplementary figure S6). Among the most dominant genera were Saccharomyces, Debaryomyces, Penicillium, Kluyveromyces and Candida. Interestingly, Saccharomyces, Debaryomyces and Kluyveromyces are found in food (cheese, bread, beer notably); they might be routinely ingested in this French cohort, suggesting a possible influence of the diet habits, although a specific study is needed to explore this hypothesis.
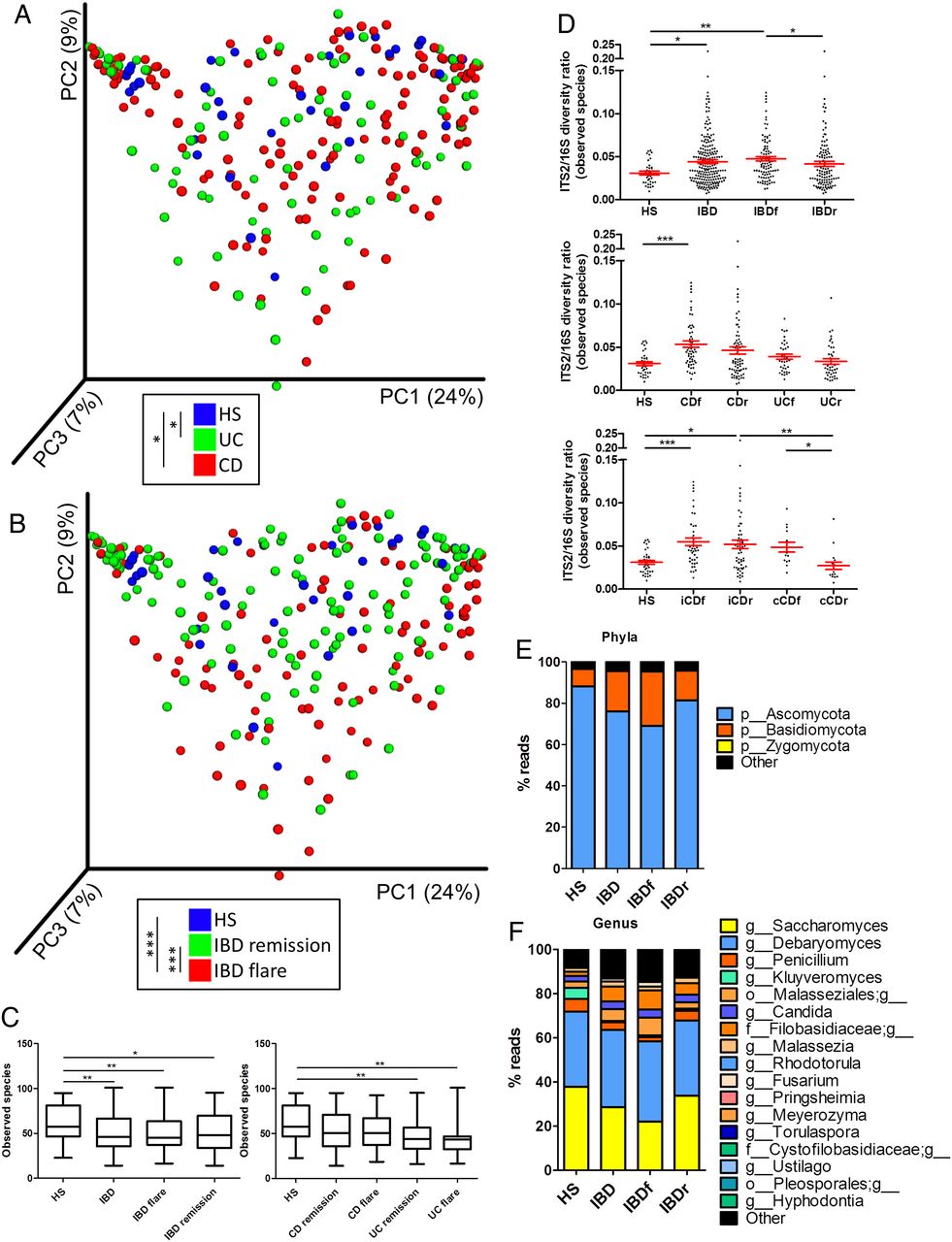
Altered fungal microbiota biodiversity and composition in IBD. (A and B) Beta diversity. Principal coordinate analysis of Bray–Curtis distance with each sample coloured according to the disease phenotype. PC1, PC2 and PC3 represent the top three principal coordinates that captured most of the diversity. The fraction of diversity captured by the coordinate is given as a percentage. Groups were compared using Permanova method. (C) Observed species number describing the alpha diversity of the fungal microbiota in the various groups studied. (D) ITS2/16S observed species ratio (Kruskal–Wallis test with Dunn's multiple comparison test. (E and F) Global composition of fungal microbiota at the phyla and genus levels. Healthy subjects (HS) and patient subgroups are labelled on the x-axis and expressed as relative OTUs abundance for each group. In all panels: *p<0.05; **p<0.01; ***p<0.001. CD, Crohn's disease.
These data show specific alterations in fungal microbiota diversity in parallel with modifications of the bacterial microbiota. Taken together, this suggests that the environmental changes during inflammation might affect differently fungi and bacteria and induce an altered fungal–bacterial inter-kingdom relationship in IBD.
Distortion in bacterial and fungal microbiota composition in IBD
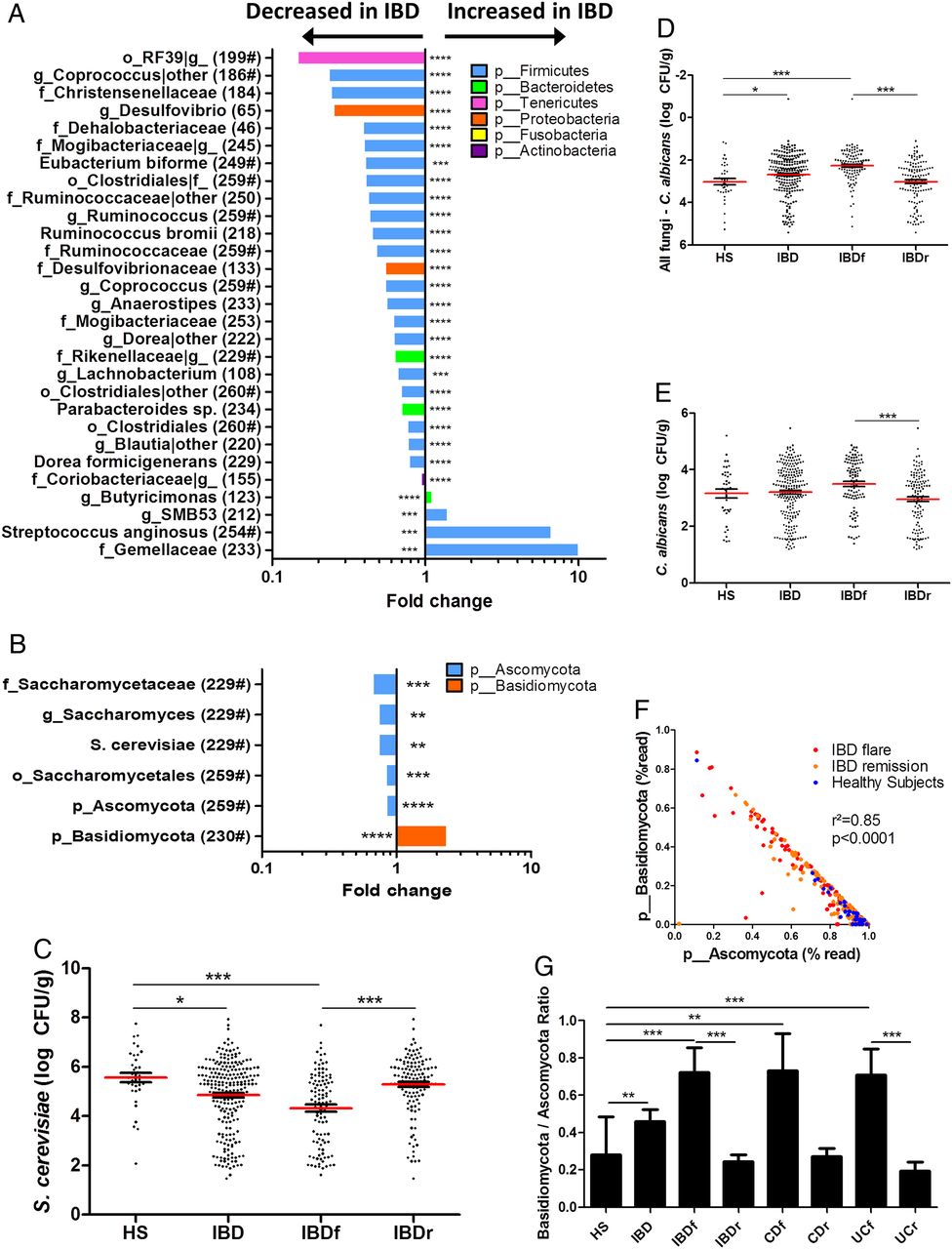
We identified the microbial features associated with disease phenotype and used a multivariate association test to control for the effects of potential confounding factors such as age, gender, smoking and treatment (multivariate association with linear models (MaAsLin); see ‘Materials and methods’ for details). Regarding bacterial microbiota, we observed an alteration in the abundance of several taxa in IBD compared with HS, in flare compared with remission and in IBD with ileal involvement compared with IBD without ileal involvement (figure 3A; see online supplementary figures S7A and 8, table S1 for full MaAsLin output). Many of these taxa have been reported in previous studies,1 ,5 including Ruminococcaceae, Lachnospiraceae, Enterobacteriaceae, Pasteurellaceae, Rikenellaceae and Gemellaceae. Notably, Ruminococcus, Coprococcus, Blautia, Eubacterium and Dorea abundance were decreased in IBD, Roseburia, Faecalibacterium, Dorea and Blautia abundance were decreased in IBD flare and Ruminococcus gnavus was increased in ileal CD. In addition to confirming these already demonstrated associations, we also found new associations such as a decrease in Anaerostipes in IBD and particularly in flare and in ileal CD. We also found an increase of Streptococcus anginosus in IBD and an increase of Aggregatibacter segnis and Actinobacillus (two members of the Pasteurellaceae family) in IBD flare compared with remission.

Bacterial and fungal taxa associated with IBD. (A and B) Differences in abundance are shown for the bacterial and fungal taxa detected using a multivariate statistical approach (see ‘Material and Methods’). The fold change for each taxon was calculated by dividing the mean abundance in the cases by that of the controls. The number of subjects that have any presence (>0) of the indicated taxon is indicated in brackets, and taxon with a mean abundance of >0.5% in at least one of the groups is indicated with ‘#’. (C) Absolute Saccharomyces cerevisiae abundance in the faecal microbiota quantified using qRT-PCR (mean±SEM). (D) Relative proportion of Candida albicans calculated by subtracting the log number of C. albicans from the log number of all fungi. (E) Absolute C. albicans abundance in the faecal microbiota quantified using qRT-PCR (mean±SEM). (F) Basidiomycota/Ascomycota relative abundance ratio in the various groups studied (Kruskal–Wallis test with Dunn's multiple comparison test, *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001). CD, Crohn's disease; HS, healthy subjects.
When analysing fungal microbiota, we identified a lower number of associations with the disease phenotype (figure 3B; see online supplementary figure S7B, table S2 for full MaAsLin output). One of the most striking features was the increased abundance of Basidiomycota in IBD and particularly in flare, which was balanced by an equivalent decrease in Ascomycota. Among the decreased Ascomycota in IBD and in flare, Malassezia sympodialis was identified. However, the Saccharomyces genus and particularly S. cerevisiae species exhibited the strongest signals. We thus performed real-time qPCR targeting S. cerevisiae on the same samples and confirmed a clear decrease in S. cerevisiae both in the absolute number and regarding the proportion in IBD and particularly in flare (figure 3C, online supplementary figure S9A-B). Although C. albicans abundance is increased in CD,14 Candida was not identified to be associated with disease phenotype in the multivariate analysis. We thus performed real-time qPCR targeting C. albicans on the same samples to specifically examine this microorganism. The C. albicans proportion and absolute number were significantly increased in IBD flare compared with IBD in remission (figure 3D, E, online supplementary figure S9C). We investigated correlations between their respective abundance. Basidiomycota and Ascomycota abundances exhibited a strong negative correlation with each other (see online supplementary figure S9D). Moreover, the Basidiomycota-to-Ascomycota abundance ratio was dramatically different according to the disease phenotype with higher values in IBD flare compared with IBD in remission and HS (figure 3F-G), suggesting that this ratio could represent a fungal dysbiosis index.
S. cerevisiae induces a regulatory response of dendritic cells
Due to the sequencing and qPCR results, we hypothesised that S. cerevisiae and C. albicans could respectively exert a protective and worsening role in the inflammatory process. As a proof of principle, we assessed the interleukin (IL)6 and IL10 production of murine bone marrow-derived dendritic cells (BMDCs) after stimulation with the two heat-killed yeast strains. The IL6 production was similar among S. cerevisiae and C. albicans; however, the production of the anti-inflammatory cytokine IL10 was significantly higher following stimulation with S. cerevisiae, suggesting an anti-inflammatory effect of S. cerevisiae compared with C. albicans (figure 4). Interestingly, the observed effects were Card9-dependent because cytokine production was nearly abolished in DC from Card9 KO mice. This confirmed the central role of this gene in host–fungi interactions.

Saccharomyces cerevisiae and Candida albicans induces distinct dendritic cell response. Interleukin (IL)10 (A) and IL6 (B) secretion by mouse bone marrow-derived dendritic cells following stimulation with S. cerevisiae and C. albicans (mean±SEM). The numbers of mice per experiment are n=5–15 (Mann–Whitney U test). KO, knockout; WT, wildtype.
IBD microbiota show-specific bacteria–fungi associations
We next assessed whether the fungi microbiota composition was correlated with the bacterial composition. To address this, we investigated a correlation at the genus level according to disease phenotype. We observed a disease-specific pattern with a higher number of significant correlations in UC compared with HS (figure 5). Although the number of correlations was similar in HS and in CD, the strength of the correlations was weaker in CD (figure 5).

Specific bacteria–fungi correlation pattern in IBD. Distance correlation plots of the relative abundance of fungi and bacteria genera. Healthy subjects (HS) (left), UC (middle) and Crohn's disease (CD) (right). Statistical significance was determined for all pairwise comparisons; only significant correlations (p value <0.05 after false discovery rate correction) are displayed. Positive values (blue squares) indicate positive correlations, and negative values (red squares) indicate inverse correlations. The shading of the square indicates the magnitude of the association; darker shades are more strongly associated than lighter shades. The sign of the correlation was determined using Spearman's method.
Interestingly, in patients with IBD, there was a positive correlation between the abundance of Saccharomyces and the abundance of several bacteria that display reduced abundance in IBD such as Bifidobacterium, Blautia, Roseburia and Ruminococcus. On the other hand, unidentified Malasseziales followed an opposite pattern. Then, to demonstrate the global intra-kingdom and inter-kingdom equilibrium according to disease phenotype, we built correlation networks at the genus level involving both bacteria and fungi (figure 6). In HS, the bacterial and fungi diversity was high (figures 1 and 2), with a network showing no marked foci of attractions with both positive and negative correlations distributed throughout the nodes. Strikingly, the CD and UC networks were dramatically different. Notably, many negative correlations connecting genera from the Proteobacteria phylum to members of the Firmicutes phylum were observed in IBD. Involvement of fungi genera from the Basidiomycota phylum, and particularly unidentified Malasseziales, in correlations was decreased in CD but increased in UC compared with HS. Similar results were obtained when Spearman's test was used to build the correlation network (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Imbalance trans-kingdom network in IBD. Correlation network in healthy subjects (A), Crohn's disease (B) and UC (C) generated using Cytoscape. Each circle (node) represents a microbial genus, its colour represents the bacterial or fungal phylum it belongs to and its size represents the number of direct edges that it has. The edge colour indicates the magnitude of the distance correlation; green indicates positive correlation and red indicates negative correlation (determined using spearman test). Statistical significance was determined for all pairwise comparisons; only significant correlations (p value <0.05 after false discovery rate correction) are displayed.
Taken together, these results suggest a complex relationship between the bacteria and fungi in the gut microbiota and that specific alterations are present in CD and UC.
Fungal microbiota–genotype association
In mice, the gut bacterial microbiota has been shown to be influenced by host genes.15 ,16 Substantial data suggest similar effects in humans.17 ,18 However, no data are available regarding fungal microbiota. We thus tested for a correlation between the relative abundance of fungal taxa associated with the disease phenotype (Basidiomycota and Ascomycota phyla as well as S. cerevisiae and M. sympodialis species) and the Card9 SNPs associated with IBD as well as several other SNPs that have been involved in defective responses to fungi.12 ,19–23 We used standard linear regression and adjusted all analyses for age, gender, smoking and treatment. With a total of 84 tests, the Bonferroni corrected p value threshold for an initial alpha of 5% was 6×10−4. As shown in table 1, which presents the 10 most significant associations, no test passed that threshold. Such a stringent significance threshold led to only limited power for our population. For example, we had 80% power to detect SNP explaining 10% (or more) of the variance of the tested taxa, but <2% power to detect SNP explaining 1% of the variance. While the presence of taxa-associated SNPs and the magnitude of the SNP effect remain to be determined, large effect size (eg, ≥10%) appear unlikely in regard of the marginal effect of these SNPs on IBD. However, we identified interesting trends: notably, a Dectin1 SNP associated with medically refractory UC (rs2078178, ‘T’ allele12) was negatively correlated with the abundance of M. sympodialis (itself decreased in IBD flare). In the same manner, the IBD-associated CARD9 SNP (rs10781499, ‘A’ allele21) was negatively correlated with the abundance of S. cerevisiae (itself decreased in IBD).
Ten most significant associations between IBD-associated fungi taxa and tested single-nucleotide polymorphisms (SNPs)
Discussion
In this study, we showed that disease-specific changes in the fungal gut microbiota are present in IBD. The simultaneous analysis of both the fungal and the bacterial microbiota enabled the elucidation of differences in the inter-kingdom equilibrium between patients with IBD and HS.
Bacterial biodiversity was decreased in both CD and UC. However, fungal biodiversity was decreased only in UC. For CD, this suggests that CD-specific environmental changes may favour fungi at the expense of bacteria. These results were particularly observed in CD patients with ileal involvement. Notably, CD patients without ileal involvement behave like UC patients with decreased fungal biodiversity (see online supplementary figure S5A, D) and only small changes in their ITS2/16S biodiversity ratio (figure 2D). Decreases in fungal gut microbiota have recently been observed in paediatric patients with IBD.24 However, the number of patients analysed in that study did not enable the authors to discriminate them according to disease phenotype and thus precluded the elucidation of CD patients’ specificities. The ileum is a major actor in intestinal physiology, notably by producing antimicrobial peptides and absorbing bile acids, two functions that are altered in ileal CD and with a potential high impact on luminal bacteria and fungi.25–28 These alterations in ileal CD could be involved in the specific microbiota alterations observed.
In accordance with published data, the fungal faecal microbiota composition of both HS and patients with IBD were dominated by fungi from the Ascomycota and the Basidiomycota phyla.7 ,24 ,29 ,30 Ascomycota and Basidiomycota abundances were strongly negatively correlated with each other and were among the most important discriminative features between IBD and HS microbiota as well as between IBD flare and remission. Logically, the Basidiomycota-to-Ascomycota abundance ratio differed between patients with IBD and HS. Compared with HS, the ratio was high in patients with IBD in flare (either UC or CD) but was normal in remission, suggesting that this imbalance between Basidiomycota and Ascomycota may be either driven by inflammation or involved in the inflammatory process.
We showed that S. cerevisiae represents a major component of the normal fungal microbiota. Its decrease, confirmed via real-time qPCR, was independently associated with IBD (vs HS) and with flare (vs remission). S. cerevisiae has recently been shown to reduce colitis induced by adherent-invasive E. coli, a bacteria associated with ileal CD, in CEACAM6-expressing mice.31 S. boulardii, a probiotic yeast closely related to S. cerevisiae,32 has been shown to exhibit anti-inflammatory effects in several colitis models7 as well as beneficial effects in the prevention of antibiotic-associated diarrhoea, acute diarrhoea, Clostridium difficile infection and enteral feeding-related diarrhoea.33 Taken together, these data suggest that S. cerevisiae could be poorly adapted to an inflammatory environment and/or that it has an anti-inflammatory potential. This raises the possibility of using fungi in new therapeutic approaches in a manner similar to what is currently being developed with several bacterial types. The results obtained using an in vitro BMDC system suggest that S. cerevisiae may exhibit regulatory effects on the host, notably by inducing IL10 production. These results are in accordance with another study reporting an increased expression of IL10 in the colons of mice receiving S. cerevisiae in a colitis setting.34 Further experiments with S. cerevisiae and other fungi, such as M. sympodialis, will be necessary to determine the role of these species in the gut and during intestinal inflammation.
Although Candida was among the most abundant genera in our studied population, it was not identified to be associated with disease phenotype in the multivariate analysis. This may be due to the high heterogeneity within the Candida genera and to the difficulty in identifying fungi at the species level using our sequencing approach. Indeed, the majority of the sequences identified as belonging to the Candida genera were not assigned to a specific species. Because several studies showed an increased level of C. albicans in patients with IBD,7 ,24 ,30 we assessed its abundance in the same samples using real-time qPCR. We confirmed a significant increase of C. albicans abundance in patients with IBD and particularly in flare.
We herein identified a disease-specific fungal microbiota dysbiosis with shifts in composition involving the two dominant fungi phyla Ascomycota and Basidiomycota and several fungi species including S. cerevisiae, M. sympodialis and C. albicans. Although changes in the fungal microbiota of patients with IBD could be connected with their altered dietary habits,35 these shifts potentially also play a role in the inflammatory process. Very little is known on the influence of M. sympodialis on intestinal inflammation. Malassezia is a genus found on the skin of mammalians and associated with numerous conditions from dandruff to atopic eczema or pityriasis.36 Despite that Malassezia clearly belongs to the human skin microbiota, species of this genus have been frequently identified in the human gut microbiota, suggesting possible colonisation of the gut.7 Indeed, the genome of Malassezia shows the presence of genes coding for secreted enzymes similar to the well-known human pathogen C. albicans. Additionally, M. sympodialis is known to secrete potent allergens that might increase local inflammation in injured part of gut of patients with IBD.37 Recent data show that M. sympodialis can activate mast cells to release cysteinyl leukotrienes and enhance the mast cell IgE response, which could contribute to inflammation.38 Moreover, we found alterations of the fungi–bacteria diversity balance in CD that might be explained by modified inter-kingdom interactions.
Fungi and bacteria coexist within the gut and may directly interact. Commensal fungi and bacteria can be found together in regions of the mouse gut.12 Antibiotics treatment in mice leads to major fungi expansions that are then reduced following antibiotic cessation,39 suggesting a balance between fungal and bacterial microbiota. In addition to differences in the ITS2/16S biodiversity ratio, we noted a disease-specific pattern for the inter-kingdom network. We performed a correlation analysis aiming at globally investigating the gut microbiota equilibrium. It showed an imbalanced microbial network in patients with IBD. In UC, the number and the intensity of the correlations between fungi and bacteria were increased. This suggested tighter interactions. On the other hand, the high ITS2/16S biodiversity ratio in CD associated with the weaker correlations between fungi and bacteria suggested a disconnection between the two kingdoms. Therefore, the role of the fungi microbiota may differ according to UC versus CD pathogenesis. In UC, the restricted biodiversity in bacteria and fungi is associated with new inter-kingdom interactions that may be involved in the inflammatory process. However, CD is characterised by disrupted connections between bacterial and fungal microbiota. This suggests that their respective effects on pathogenesis may be independent. Further studies are needed to elucidate more precisely the functional connections within and between kingdoms in the gut microbiota.
Finally, although our study was not of sufficient power to statistically demonstrate an association between genotype and fungal microbiota, we identified some trends suggesting that SNPs associated with IBD or IBD severity may influence fungal microbiota dysbiosis.
Conclusion
In addition to elucidating bacterial dysbiosis, our study identified a distinct fungal microbiota dysbiosis in IBD that is characterised by alterations in biodiversity and composition. Moreover, here, we unravel disease-specific inter-kingdom network alterations in IBD, suggesting that, in addition to bacteria, fungi may also play a role in IBD pathogenesis. Identifying the key players of these inter-kingdom interactions and understanding how they influence or are influenced by gut inflammation are further research avenues to pursue.
Materials and methods
Patients and samples collection
All patients were recruited at the Gastroenterology Department of the Saint Antoine Hospital (Paris, France) and provided informed consent. A diagnosis of IBD was defined according to clinical, radiological, endoscopic and histological criteria. None of the study participants had taken antibiotics or used colon-cleansing products for at least two months prior to enrolment. Patient characteristics are presented in online supplementary table S1. Faecal samples were collected from 235 patients with IBD and 38 HS. Whole stools were collected in sterile boxes and immediately homogenised, and 0.2 g aliquots were frozen at −80°C for further analysis.
Microbiota analysis in healthy subjects and patients with IBD
Faecal samples were subjected to DNA extraction as previously described.3 The DNA samples were then used for 16S and ITS2 gene sequencing and sequence analysis (see online supplementary materials and methods). For specific analysis of C. albicans and S. cerevisiae population, real-time quantitative PCR on the faecal DNA was done (see online supplementary materials and methods).
In vitro experiments on the response of dendritic cells to S. cerevisiae and C. albicans
BMDCs were generated as described in the online supplementary materials and methods. BMDCs were stimulated with heat-killed C. albicans or S. cerevisiae at an multiplicity of infection (MOI) of 10 during 18 h and the cell culture supernatant was used for ELISA analysis (see online supplementary materials and methods).
Genotyping is described in the online supplementary materials and methods.
Statistical analysis
GraphPad Prism V.6.0 (San Diego, California, USA) was used for all analyses and graph preparation. For all graph data, the results are expressed as mean±SEM, and statistical analyses were performed using the two-tailed non-parametric Mann–Whitney U test or Kruskal–Wallis test with Dunn's multiple comparison test. Statistical significance of sample grouping for beta diversity analysis was performed using Permanova method (9999 permutations). Differences with a p value <0.05 were considered significant.
MaAsLin, a multivariate statistical framework, was used to find associations between clinical metadata and microbial community abundance.14
Correlation within microbial taxa abundance data was measured by correlation proposed by Szekely and Rizzo40 and recommended by Simon and Tibshirani.41 It was used with success in other microbiota data analysis42 as well as in human genetic to identify gene network.43 It is scale free and allows detecting non-linear relationship. The distance correlation is bounded by 0 and 1 and is equal to 0 if there is independence between variables. A statistical test is provided to assess for the dependence between variables and is shown powerful and easy to compute.41 In addition to the distance correlation, the Spearman's correlation sign was computed to describe heuristically the direction of association between microbial taxa. The distance correlation was computed under R-3.2.3 using the package energy v1.6.2. The p values were corrected using Benjamini and Hochberg to control false discovery rate. Correlation networks were built using Cytoscape 3.1.1.
To investigate correlations between fungi taxa and genotype, we performed a systematic association screening between the four major independent taxa that were previously identified to be associated with IBD (vs HS) or with flare (vs remission) (figure 3B, online supplementary figure S7B) and the 21 common candidate SNPs. All outcomes were standardised using a rank normal transformation using the function rntransform from the R package GenABEL ( http://cran.r-project.org/web/packages/GenABEL/index.html). We used standard linear regression and adjusted all analyses for age, gender, smoking and treatment.
Acknowledgments
The authors thank Christophe Hennequin from CH Saint Antoine for providing them with the S. cerevisiae strain. They also thank Elodie Drouet and the Clinical Research Assistant team of Unité de Recherche Clinique de l'Est Parisien (Assistance Publique–Hôpitaux de Paris and UPMC Paris 06) for their help in obtaining the IBD patient samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online tables
Footnotes
Contributors HS conceived and designed the study, performed data analysis and wrote the manuscript. VL and SJ conducted all experiments unless otherwise indicated below. HA performed the statistical analysis of genetic data. H-PP performed the statistical analysis of the pyrosequencing data. DC and MLR performed the in vitro experiments. CL, AB, IN-L, GL, JC, PS, PL, DS, MLR, LB, and HS discussed the experiments and results.
Funding This study was supported by the Broad Medical Research Program, the INSERM cohort grant and the Association Francois Aupetit.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Local ethics committee of Comite de Protection des Personnes Ile-de-France IV, IRB 00003835, Suivitheque study, registration number 2012/05NICB.
Provenance and peer review Not commissioned; externally peer reviewed.