Article Text

Abstract
The microbiome has received increasing attention over the last 15 years. Although gut microbes have been explored for several decades, investigations of the role of microorganisms that reside in the human gut has attracted much attention beyond classical infectious diseases. For example, numerous studies have reported changes in the gut microbiota during not only obesity, diabetes, and liver diseases but also cancer and even neurodegenerative diseases. The human gut microbiota is viewed as a potential source of novel therapeutics. Between 2013 and 2017, the number of publications focusing on the gut microbiota was, remarkably, 12 900, which represents four-fifths of the total number of publications over the last 40 years that investigated this topic. This review discusses recent evidence of the impact of the gut microbiota on metabolic disorders and focus on selected key mechanisms. This review also aims to provide a critical analysis of the current knowledge in this field, identify putative key issues or problems and discuss misinterpretations. The abundance of metagenomic data generated on comparing diseased and healthy subjects can lead to the erroneous claim that a bacterium is causally linked with the protection or the onset of a disease. In fact, environmental factors such as dietary habits, drug treatments, intestinal motility and stool frequency and consistency are all factors that influence the composition of the microbiota and should be considered. The cases of the bacteria Prevotella copri and Akkermansia muciniphila will be discussed as key examples.
- obesity
- diabetes mellitus
- intestinal microbiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Microbes that reside in the human gut are key contributors to host metabolism and are considered potential sources of novel therapeutics. Although this sentence may be seen as obvious in 2018, the universality of this concept is less evident. Undeniably, it is due to the advent of genetic tools and the metagenomic revolution of the last 15 years that we are now able to characterise the composition and function of microbiomes (box 1) from different parts of the body and link them to potential diseases, risks or even to the clear onset of clinical symptoms. In recent decades, microbes have mostly been used to develop disease-specific diagnostics. Currently, the mechanisms of interactions or of defense against potential pathogens are often described at the molecular level. Moreover, the current understanding is that some gut bacteria may also achieve this goal by communicating with human cells and mostly by promoting immune interactions.1 2 A large number of recent papers and reviews have covered different aspects of the microbiome and its potential role in human health, including the early life3–5 but also specific diseases, such as cardiometabolic disorders, inflammatory bowel diseases, neuropsychiatric diseases and cancer.6–12
In the present perspective review, recent evidence showing the impact of specific bacteria and the involvement of the innate immune system will be discussed. However, most importantly, we debate one part of the current knowledge in this field and ask the following question: how should we interpret the numerous hopes, promises and threats?
Before discussing specific examples, we recommend that you scrutinise the next paragraph, which introduces us to the infinite minuscule world that may have great impact on human health (figure 1).

Relative sizes of major host cells and their components versus those of bacteria and viruses.
The gut microbiota is now considered an important partner of human cells, interacting with virtually all human cells. In 2017, approximately 4000 papers focusing on the gut microbiota were published , and between the years 2013 and 2017, more than 12 900 publications were devoted to the study of the gut microbiota. This remarkable number represents more than 80% of the overall publications of the last 40 years (since 1977) on this topic. Therefore, this simple finding highlights the fact that this field of research is not only blossoming but also strongly suggests the necessity for advancement.
Although not discussed in detail on this perspective review, we should briefly mention that in addition to bacteria, other key microorganisms, such as archeae, viruses, phages, yeast and fungi, are present in the gut. These microorganisms, which likely control the activity of the host and, most importantly, of the gut microbes, have been investigated in detail and may be as important as bacteria. Therefore, the archaea, the virome, the phageome and the mycobiome offer an additional dimension to the investigation of host–microorganism interactions. As an example, the phages do not only exceed the number of bacteria (eg, 10-fold more phages than bacteria) but they are also new actors playing roles in these complex interactions.13–17 As a simple example, it has been more than a 100 years since Félix d’Herelle formally identified phages15 (figure 1). However, reproducible protocols to analyse the faecal phageomes using metagenomics analysis have only recently emerged.18 Therefore, this implies that this area of research requires more time before major fundamental breakthroughs can be translated to general applications for the public. Nevertheless, today, due to the different kind of media (ie, mode of communication), medical information can be rapidly disseminated to the public. Hence, one of the major caveats remains the lack of perspective not only from society but also from some scientists and healthcare professionals, who may misinterpret data or are expecting a direct translation of such complex research from the bench to the clinic. Therefore, experts in this field must provide access to such knowledge with caution and without delusion.
Microbes interact with host cells: immunity links microbes to metabolism
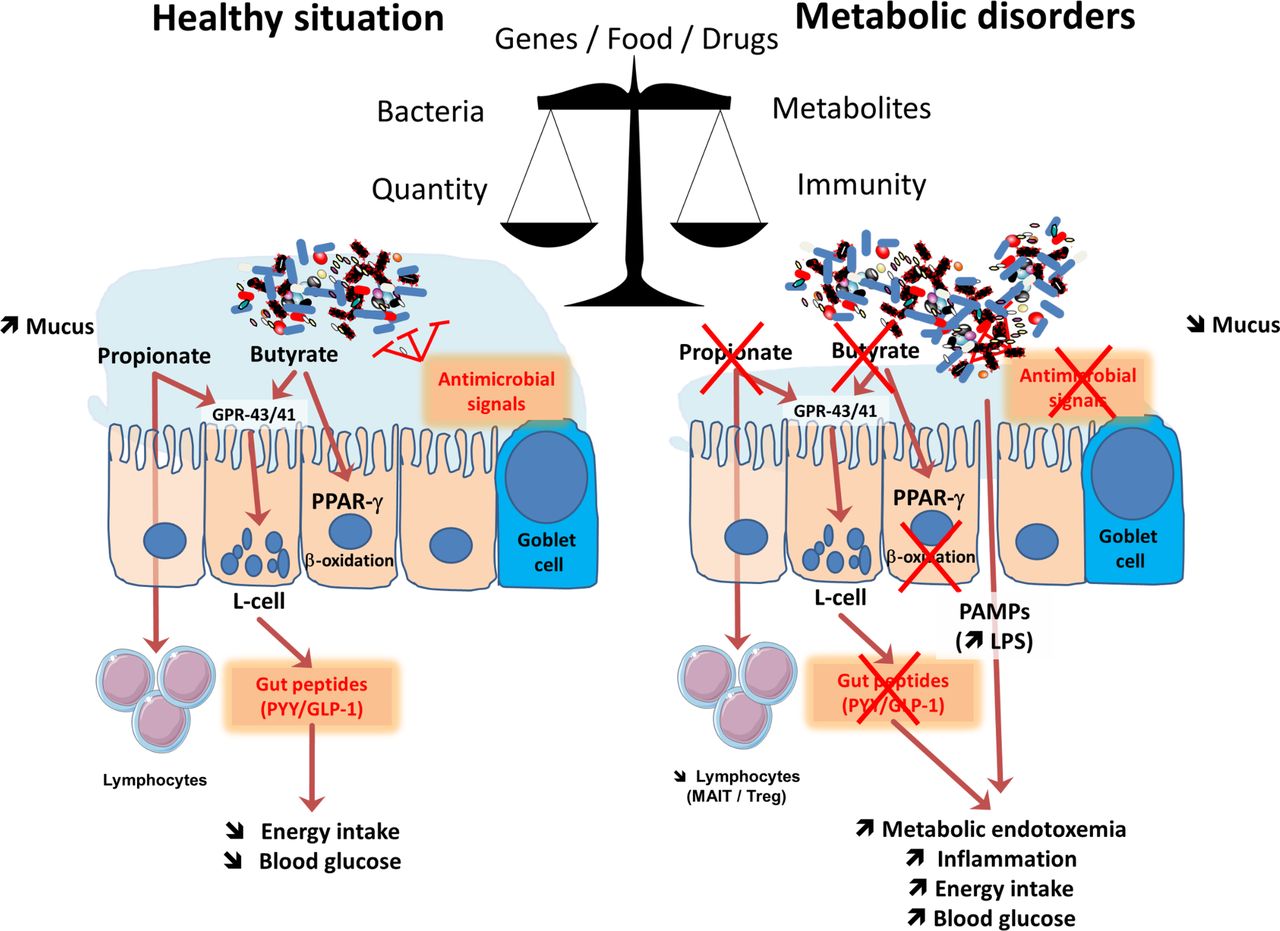
Different systems recognise and monitor the presence of microorganisms in the body. For example, in the gastrointestinal tract (GI), epithelial cells play a major role as guardians that translate key information to the immune cells located in the lamina propria. In fact, the recognition and the monitoring of microbes is mainly performed by the innate immune system with pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) and NOD-like receptors (NLR).19 The TLRs are transmembrane receptors expressed on cell surfaces (ie, TLR-2, 4, 5) or in endolysosomal compartments (ie, TLR-3, 7, 8, 9 or 13), whereas NOD receptors are cytosolic proteins. Together, these receptors recognise pathogen-associated molecular patterns (PAMPs) from microorganisms (eg, lipopolysaccharides, peptidoglycans, lipoteichoic acid, flagellin and muramyldipeptide) or danger-associated molecular patterns from damaged tissues.19 Thus, the GI tract not only contains a vast majority of the microbes that reside in the human body but also harbours probably the larger pool of the immune cells that are present in the body.20 Notably, the immune system also has a predominant effect on the composition of the microbiota.21 Therefore, GI cells are continually exposed to a vast number of microbial antigens and metabolites. In spite of this close proximity, we live with microorganisms in perfect symbiosis (figure 2).

Major mechanisms involved in the crosstalk between microbes and host: impact of metabolism. The balance between healthy and pathological situations (eg, metabolic disorders) is crucial. This is under the tight influence of several factors including the genes, food and drugs. This left part of the figure shows that in healthy situation, the composition of the gut microbiome is associated with a higher mucus layer thickness, the production of antimicrobial signals and different short-chain fatty acids such as butyrate and propionate. Both butyrate and propionate bind to G protein coupled receptors (GPR)-43 and GPR-41 expressed on the enteroendocrine L-cells thereby stimulating the secretion of gut peptides such as glucagon-like peptide-1 (GLP-1) and peptide YY (PYY). This effect contributes to reduce food intake and to improve glucose metabolism. Propionate can also bind to GPR-43 expressed on lymphocytes in order to maintain appropriate immune defence. Butyrate activates peroxisome proliferator-activated receptor-γ (PPAR-γ) leading to beta-oxidation and oxygen consumption, a phenomenon contributing to maintain anaerobic condition in the gut lumen. As depicted on the right part of the figure, during metabolic disorders, changes in the gut microbiome are linked with a lower mucus thickness, decreased antimicrobial defense and butyrate and propionate production. As a consequence, L-cells secrete less gut peptides. The lack of PPAR-γ activation lead to higher oxygen available for the microbiota at the proximity of the mucosa and increases the proliferation of Enterobacteriaceae. The decrease in propionate also contribute to the lower abundance of specific T cells (mucosal-associated invariant T cells (MAIT) and Treg) in the lamina propria of the gut. Altogether, such changes in the microbial environment and metabolites induce a leakage of pathogen associated molecular patterns (PAMPs) such as the lipopolysaccharide (LPS) that are increased in the blood, and trigger low-grade inflammation.
In addition to the classical immune considerations, the interactions between gut microbes and the immune system have led to the discovery of previously unknown functions; for example, it was discovered that specific microbial components strongly contribute not only to the regulation of energy metabolism but also to glucose and lipid homeostasis.22–24 In 2007, we first identified that constituents of gram-negative bacteria, such as lipopolysaccharides (LPS), were the key factors triggering the onset of low-grade inflammation and insulin resistance via mechanisms of interaction between gut microbes and the innate immune system (ie, TLR-4, CD14).22 Indeed, genetic models of both diet-induced obesity and diabetes were characterised by an increased level of circulating LPS, a condition called metabolic endotoxaemia,22 which was later confirmed in humans (figure 2)25–34. Since the initial identification of LPS as a major actor involved in the onset of metabolic alterations associated with overweight and obesity (eg, insulin resistance, glucose intolerance, dyslipidaemia, hepatic steatosis), other PAMPs such as peptidoglycans or flagellin have been shown to play a causal role on the regulation of similar metabolic pathways.35–37
In addition to the specific changes in the composition of the gut microbiota, it is now accepted that several key factors contribute to the translocation of bacterial compounds from the intestinal lumen to the body. As previously indicated, gut microbes are constantly interacting with intestinal epithelial cells; however, in normal conditions, the gut barrier function is highly efficient due to complex multifaceted mechanisms (ie, tight-junction proteins, thickness and composition of the mucus layer, presence of antimicrobial factors, intraepithelial lymphocytes and other adaptive immune cells and production of immunoglobulin A (IgA)) (figure 2) (reviewed in König et al and Wells et al 38 39).
It is also well known that a loss of immune tolerance is associated with bowel inflammation. Interestingly, recent data have also shown the accumulation of T cells in the intestines of obese subjects consuming high-fat diets, an observation that correlates with morbidity.40 Conversely, the circulation of other immune cells, such as mucosal-associated invariant T cells (MAIT) (ie, innate-like T cells), which exhibit elevated Th1 and Th17 cytokine production is strongly decreased in patients with obesity and type 2 diabetes41 (figure 2).
In conclusion, the gut barrier is controlled by fine-tuned communications occurring between gut microbes and the host immune system. Additionally, the complexity of those interactions raise the question about the level of our current understanding and eventually contribute to explain why it is relatively difficult to develop specific therapeutic targets.
Microbial activity, metabolites and metabolism
In addition to receptors recognising specific constituents of the cell membranes of microbes and directly related to the innate immune system, there is evidence that many different microbial metabolites also influence host metabolism mostly by binding to specific host membranes or nuclear receptors (for review see Husted et al, Pallister et al, Brown and Hazen, Rastelli et al, O’ Mahony et al, Kasselman et al 42–47). Among the numerous metabolites produced by the microbiota, we can mention important metabolites such as folate, indoles, secondary bile acids, trimethylamine-N-oxide (TMAO), but also neurotransmitters (eg, serotonin, gamma amino butyric acid), and eventually short-chain fatty acids (SCFAs). The latter example is likely the most studied. Indeed, the most investigated are probably the SCFAs (ie, butyrate, propionate and acetate). It has been shown that SCFAs are recognised by G-protein-coupled receptors such as GPR-41 and GPR-43.48 The stimulation of these receptors triggers the secretion of intestinal peptides involved in glucose metabolism or food intake, such as glucagon-like peptide-1 or peptide YY (PYY)49 50 (figure 2). Therefore, by stimulating enteroendocrine cells to produce key hormones, microbes act from a distance on different organs.51–53 Notably, propionate also regulates immune cells to produce antimicrobial factors and, therefore, may act as an immune regulator,54 including a reduction of cancer cell proliferation.55 56 This latter example also highlights the fact that a distinct microbial metabolite may play different roles, ranging from the regulation of glucose levels to immunomodulatory effects, in host metabolism.
Along these lines, in reality, the role and mechanisms of action of some SCFAs might also be very different from what has previously been thought. For example, butyrate has been considered for decades to be an essential energy source, allowing colonic cells to proliferate and contributing to the maintenance of healthy gut barrier function. However, in a recent study, Byndloss et al discovered that butyrate may also strongly influence the microbial environment and ecology by communicating with host cells. It has been shown that butyrate instructs colonic cells to ‘breathe’ oxygen by activating β-oxidation in order to protect the host against the expansion of potentially pathogenic bacteria to the intestinal lumen (figure 2).57 58 Specifically, the authors used recent information suggesting that the extremely low quantity of oxygen present in the luminal content of the gut (ie, the anaerobic state) is a condition that is required to prevent the expansion of putative facultative anaerobic pathogens such as Salmonella or Escherichia.59 Thus, the authors demonstrated that the consumption of oxygen by the host cell to β-oxidise butyrate in the mitochondria contributes to limiting the diffusion of oxygen from the colonic cells into the luminal compartment, eventually leading to the maintenance of anaerobic conditions57 58 (figure 2).
What matters: quality, quantity or activity of microbes?
Over the last decade, pioneering papers have shown that some metabolic disorders, such as obesity and diabetes, are associated with shifts in the microbiota at the phylum level (ie, in the Firmicutes/Bacteroidetes ratio).60 Since this discovery, not all papers have been able to replicate this finding, which gives rise to the following question: should we focus only on the general composition at the phylum level or should we go deeper (ie, the genus and species levels)? However, in addition to these considerations, there is another important question: is it more relevant to explore the metabolic capacity of the intestinal microbiota and, eventually, to the metabolites produced (eg, butyrate, bile acids, TMAO) than it is to study the microbial composition alone?42 51 61 62 Indeed, as briefly described in the previous chapter of this perspective, there are numerous metabolites produced by the gut microbiome that can influence our metabolism.
At this stage of our knowledge, there are no clear answers to these questions. Along these lines, in addition to these discoveries and observations, a recent, very elegant study demonstrated that it is the absolute quantity of microbes and not the proportions of microbes that really matters.63 This study strongly argues that most previous studies, which have primarily been based on the investigation of the relative proportions of microbes, have probably not been able to capture the essence of the problem. For example, the authors observed differences of up to 1 log between the microbial loads of healthy individuals. As another striking example, in this paper, the authors highlighted the fact that the abundance of Bacteroides is associated with Crohn’s disease only when using the classical relative abundance-based profiling, whereas by using quantitative microbiome profiling, it was the abundance of the genus Prevotella that was found to be decreased in patients with Crohn’s disease.63 Moreover, this observation underlines the real risk and the limitations of using relative abundance-based analysis because this type of analysis can lead to erroneous interpretations (figure 2).
Similarly, the necessity for analysing not only the presence of certain gut microorganisms but also the activity of these organisms has already been discussed by authors.64 65 More recently, Schirmer et al profiled the gut microbiomes of more than a hundred individuals, including patients with Crohn’s disease, ulcerative colitis and non-IBD control patients and analysed both the metagenomes and the metatranscriptomes of these individuals.64 They discovered that most of the bacteria were well correlated when analysing DNA and RNA abundances; however, strikingly, some organisms that were abundant in terms of metagenomic data were, as stated by the researchers, ‘inactive or dormant’ in the gut with little or no expression. Conversely, some disease-specific bacterial characteristics were only detectable when analysed at the transcriptional level. Therefore, such findings highlight the fact that the transcription of specific gut microbial pathways may vary over time, leading to potential phenotypic changes that may be complementary to changes associated with classical metagenomic abundance (figure 2).
Having said that, the specific examples highlighted above and relating the quantity of bacteria and the activity of the bacteria (including the metabolome) strongly demonstrate the importance of multiomics approaches and how various genomic and metabolomics approaches complement each other to further dissect the interactions between microbes, host and the overall metabolism.66
Several interesting studies have also proposed that the microbiome should be seen as an integral part of precision medicine approach (for review see Suez and Elinav and Kashyap et al 67 68) because it contributes to interindividual variability in all aspects of diseases situation but is also a modifiable factor leading to the development of future therapeutics. For example, Zeevi et al elegantly illustrate this concept by showing that the gut microbiome can be used to predict personalised blood glucose responses to specific diets, which differ between individuals.69 However, this has required the use of machine-learning algorithm that integrates many parameters, such as in this case the week-long glucose levels, dietary habits, anthropometrics, physical activity and gut microbiota from more than 800 individuals and more than 45 000 meals. Interestingly, the algorithm was then tested in an independent cohort of 100 individuals.69 Although found as highly promising and clearly showing the application by some researchers, others have requested to tone down the potentiality of a broad applicability with conclusions such as the fact that predictive algorithms are black boxes with complex statistical associations without the real mechanisms explaining the presence of such associations.70 Or others mentioned that the results does not provide enough evidence that the model is finally superior to the current methods of detecting high glucose levels and even did not demonstrated that personalised nutrition advice are superior to standard advices in view of managing high glucose levels in postprandial situations.
Actually, there is not yet a full consensus reached on what is the best option and how to proceed to design future personalised medicine, but the field is relatively young and warrants further work.
Nevertheless, obtaining a more comprehensive analysis of the situation by screening not only the composition but also the metabolites (eg, metabolome) in prospective conditions (as opposed to the current conditions that have the caveat of only studying single time points) will likely be useful. Notably, one may also argue that most studies have examined faecal material (which is easily accessible), whereas the mucosal microbiota is still under investigated (figure 2). This without saying that such ambitious studies will require standardised protocols for sampling, storage and analysis. In addition, as it has been done for many years in the area of nutrition and drug treatments, we should probably develop large-scale longitudinal studies. Indeed, these kind of future studies should be designed to allow a follow-up of several years in a very broad cohort of subjects (ie, apparently healthy as well as diseases subjects) during which samples (eg, blood, urine, faecal) as well as many as possible information should be carefully followed on top of the classical dietary habits and drug use.
Altogether, the different points discussed above strongly support the need for functional studies but it also highlight the current difficulties to obtain a clear consensus on the best way to proceed. Hence, to obtain a complete overview of the role of the microbiome on health, we will still need to combine compositional analysis of microbiomes and showing changes correlating to environmental changes (ie, diets, drugs) or biological states (ie, interindividual physiological variations, diseases). This will then require follow-up studies to validate the causality. This is the purpose of the next chapter of this perspective review.
Hopes, promises and threats: the indispensable need for proofs of concept
This last part of the review is dedicated to a discussion of critical issues and the necessity to develop well-defined experiments before claiming that some microbes are or not beneficial or even deleterious. As stated above, when a correlation is found between a given microbe and a disease or healthy situation, it is challenging to show the exact implication of the candidate on the onset of the disease or conversely its beneficial impact.
In other words, as mentioned in the previous paragraphs, the literature often discuss the role of specific candidates (ie, bacteria) as potential beneficial or deleterious candidate following correlative works. However, sometimes we probably rapidly (too rapidly) infer positive or negative effects without properly investigating these effects.
Therefore, we, as researchers usually start from correlation and seldom provide proof of concept by using isolated bacteria or identified metabolites. This is not because we do not want to investigate properly the roles of ‘new’ microbes or metabolites; but because it might be difficult to culture some bacteria. Indeed, moving from the signature identified at the 16S level to the proper isolation of the candidate bacteria and to investigations in complex models (even in vitro) usually requires much time and effort. Although recent advances in culturomics have been made,71 72 the isolation and the identification of anaerobic bacteria remain time consuming and fastidious.72 73 Moreover, when a putative candidate has been isolated and cultured, it is still necessary to culture it in quantities that are compatible with chronic in vivo testing.
On top of that, at the level of our knowledge, it is still very difficult to fully decipher the role of any microorganism in a complex community such as the gut microbiota. This last point is illustrated by two specific examples: the cases of two bacteria that is Prevotella copri and Akkermansia muciniphila.
The case of P. copri
The role of P. copri has been recently investigated in two independent elegant studies. Although, both seminal papers reached their respective conclusion from data obtained from human observational studies and subsequently confirmed in mouse intervention studies, they represent a clear example on how misleading a simple association study can be and how potential factors may influence discrepant and even contrasting results from functional proof-of-concept studies.
In the first one, P. copri was found to improve glucose metabolism and insulin sensitivity by a mechanism associated with the production of succinate on the fermentation of dietary fibres.74 More precisely, De Vadder et al have discovered that a diet rich in fermentable dietary fibres was associated with a higher production of succinate.74 They found that the abundance of succinate was higher in the caecal content but not in the portal vein of the mice treated with dietary fibres. Therefore, they hypothesised that succinate was used as a substrate by the intestinal cells. Accordingly, succinate is a substrate for the intestinal gluconeogenesis, a process well described to improve glucose homeostasis.75 More importantly, they demonstrate that succinate feeding improves glucose tolerance and insulin sensitivity. Similarly, the colonisation of mice with P. copri replicates this beneficial phenotype in high-fat/high-sucrose fed mice.74 Finally, by using a mouse model with an intestinal epithelial cell-specific deletion of the rate-limiting enzyme involved in the gluconeogenesis (ie, glucose-6-phosphatase), they demonstrated that succinate produced by P. copri is an essential mechanism involved in the improvement of glucose and insulin sensitivity.74
In the second study, Pedersen et al discovered that the metabolome of insulin-resistant individuals is characterised by the presence of high levels of branched-chain amino acids (BCAAs) which correlates with a microbiome enriched in the biosynthetic pathways of such BCAAs.76 Among the bacteria linked with this observation, both P. copri and Bacteroides vulgatus were identified. To experimentally address the relation between P. copri and an altered glucose metabolism, they gavage high-fat fed mice with P. copri or a vehicle. The authors discovered that P. copri administration aggravates glucose intolerance, reduces insulin sensitivity and increases serum total BCAAs level.76
In conclusion, although both studies are designed and performed as proof-of-concept studies, whether P. copri should be considered as a beneficial or deleterious bacterium likely depend on the dietary environments and probably requires further investigations.74 76
Another key recent example is the large amount of interest generated by the bacterium A. muciniphila. This bacterium is one of the most abundant single species in the human intestinal microbiota (up to 5% of the total bacteria in basal conditions) and was isolated and characterised as a mucin-utilising specialist by Professor Willem de Vos in 2004.77 Today, A. muciniphila is viewed by several scientists as a next-generation beneficial microbe.78–81
The case of Akkermansia in cardiometabolic disorders
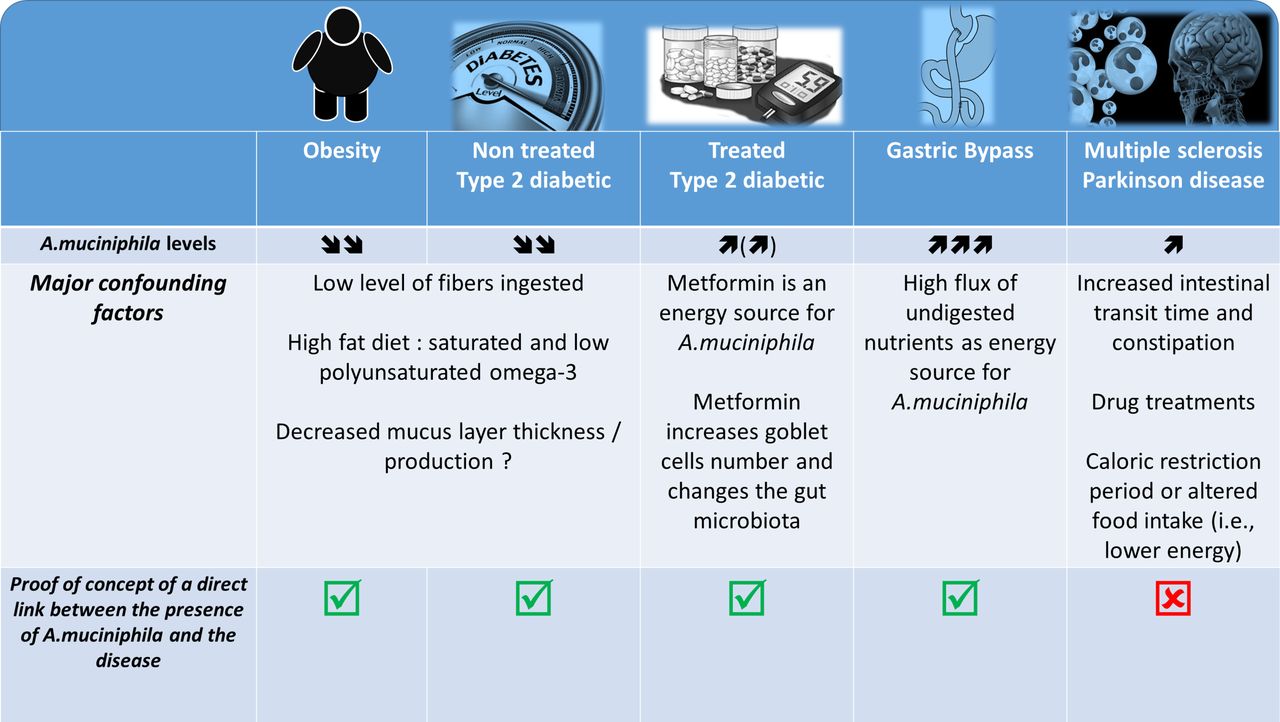
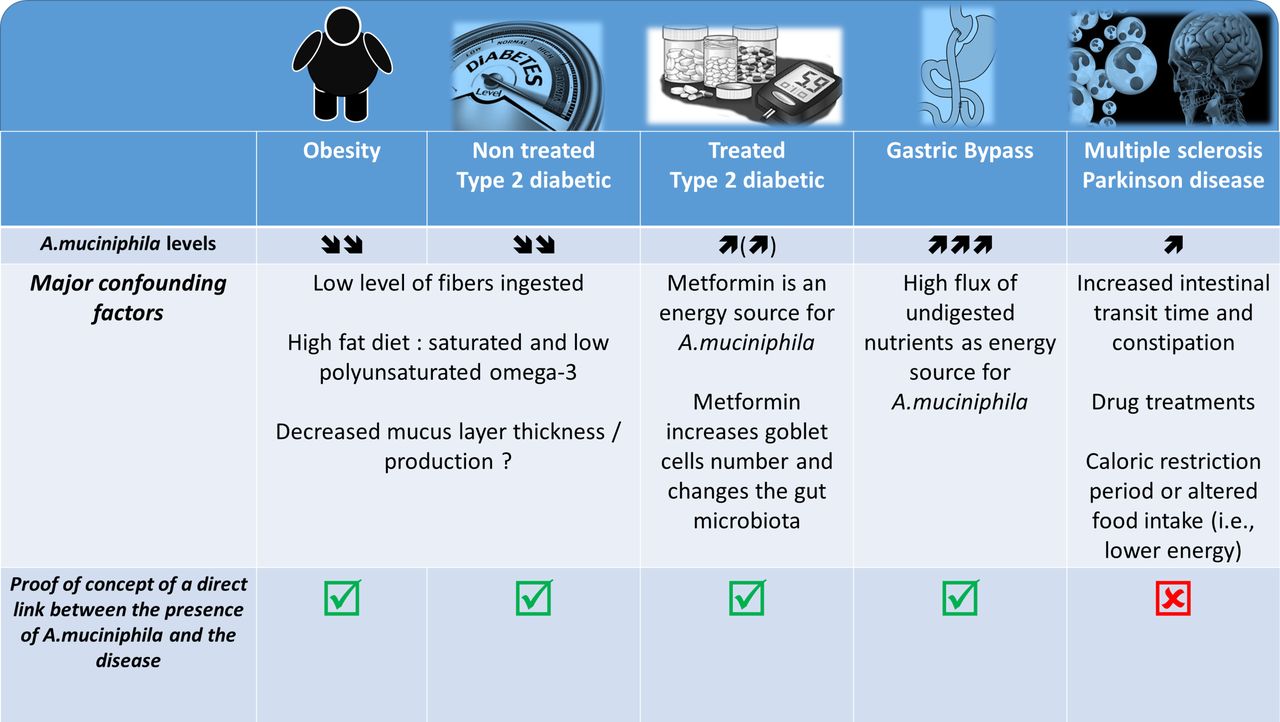
It was described that A. muciniphila was less abundant in the intestinal microbiota of genetically obese and diabetic mice as well as of mice in which these conditions were dietarily induced. Interestingly, in these mice, the abundance of A. muciniphila was systematically found to be inversely correlated with body weight, fat mass, inflammation, insulin resistance and glucose intolerance (figure 3).82–89 This observation therefore suggested that this bacterium might be of interest. However, few other studies in mice have reported an increased abundance of A. muciniphila on the ingestion of a high-fat high-sucrose diet.90 91 Conversely, it has also been widely demonstrated that prebiotic feeding (eg, with inulin-type fructans and some polyphenols) strongly increases the presence of A. muciniphila and improves metabolic disorders associated with obesity, including decreased fat mass, insulin resistance, lower liver steatosis and reinforcement of the gut barrier.82 85 91–93 Importantly, in humans, the abundance of A. muciniphila was seen to be decreased in several pathological conditions, such as obesity, type 2 diabetes, hypertension, hypercholesterolaemia and liver disease94–100 (figure 3). Metformin, which is the most extensively used antidiabetic treatment, and bariatric surgery dramatically increased the abundance of A. muciniphila (up to 20% of the total microbiota).101–104
{kind=link}
{kind=link}
{kind=link}
Association between Akkermansia muciniphila and several diseases: what is known? What are the major confounding factors. The picture illustrated different pathological situations where the abundance of the bacteria A. muciniphila has been found to be increased or decreased. It also highlight several confounding factors associated with the modulation of the gut microbiota and eventually the abundance of A. muciniphila according to the health situation and shows the current data for which a proof-of-concept of the link between the disease and the presence of the bacteria has been made.
In addition to these data, which are abundant but correlative, over the last 2 years, several papers have adopted a proof-of-concept strategy by investigating the impact of the A. muciniphila administration in vivo and in different models. It is now accepted that supplementation with A. muciniphila protects against several cardiometabolic features. All the studies in which animals were treated with A. muciniphila showed that the bacteria lowers body weight and fat-mass gain, hepatic steatosis, inflammation, cholesterol levels and atherosclerosis; improves insulin sensitivity and restores gut barrier function by influencing different factors (ie, mucus-layer thickness, tight-junction proteins, antimicrobial peptides and immunity)83 96 105–108 (figure 3). Notably, one of the potential key mechanisms by which A. muciniphila acts is via specific compounds such as the protein ‘Amuc_1100’.105 Recently identified on the outer membrane of this bacteria, this protein has been shown to play immunomodulatory roles both in vivo and in vitro,105 109 and this effect of A. muciniphila on the immune system is important because A. muciniphila has recently been shown to delay the onset of type 1 diabetes in diabetes-prone animals.107
Akkermansia and immune-based cancer therapy: proof of concept
With regard to immunity, the impact of A. muciniphila seems to be of great importance not only in the context of metabolic syndromes or for reducing autoimmune type 1 diabetes as described above105 107 but also in the context of cancer therapy. It has been previously shown that anticancer therapies may rely on the composition of the gut microbiota.110 However, in a series of recent studies,111–113 a role for A. muciniphila in this context has been discovered. Specifically, the authors have shown that the gut microbiota and even specific key bacterial species may influence the outcomes of anticancer immunotherapy, such as anti-PD-1 treatment.111–113 First, the authors simply observed that the patients responding to anti-PD-1 treatment were those exhibiting increased abundance of gut A. muciniphila compared with non-responders. This observation suggested that a favourable drug response might be observed in the presence of A. muciniphila. While this finding was a simple correlation, the authors clearly moved to a proof-of-concept strategy when they decided to transfer into mice the microbiota from responders and non-responders. In this experiment, the authors observed a similar response, that is, an altered drug response in the absence of A. muciniphila. Although they used particular models, that is germ free or antibiotic-treated mice colonised with the microbiota from responder or non-responder donors, the treatment with the bacterium A. muciniphila reversed the compromised efficacy of anti-PD-1 blockade in mice receiving the microbiota from non-responders. Therefore, poorly responding mice (ie, colonised with non-responders faecal material) turned into responders on treatment with A. muciniphila, clearly showing that the presence of this bacteria plays an important role. More importantly, the authors found that this result was the consequence of an immunomodulatory effect of A. muciniphila.111 Despite this very important finding, we may not rule out that the effects are not directly mediated by this bacterium but requires the activity (or cross-talk) with one or several other key species which drive the beneficial effects. Moreover, further human studies are needed in order to consider the bacteria as therapeutic adjuvants in this context.
The case of Akkermansia in neurodegenerative disorders
While different proof-of-concept studies have been described above, there are other situations where the over-representation of A. muciniphila in the faeces are not associated with a beneficial effect. In fact, a series of papers investigating Parkinson’s disease and multiple sclerosis (MS) have been recently published.114–116
More specifically, recent papers have characterised the gut microbiome of patients with MS and have identified variations in specific microbes. Indeed, the levels of 25 different bacteria were significantly different between diseased patients and controls, and one of these bacteria was A. muciniphila.114 115 Strikingly, the studies by Berer et al and Cekanaviciute et al focused their attention on the increased abundance of A. muciniphila in the faecal material of patients with MS. However, although both studies elegantly demonstrated that the faecal microbiota of MS-affected individuals worsened disease progression compared with control individuals when transplanted into a mouse model of MS, none of these studies was able to detect the presence or any change in the abundance of A. muciniphila after the transplantation of the microbiota from the human donor into the recipient mice. Consequently, this observation exclude at this stage of the study the role of A. muciniphila in the onset of the disease.
This finding also suggests that the presence of A. muciniphila in the human stool samples but not in the rodents that developed the disease by being colonised with the same human faecal material may be due to other bacterial candidate. Along this line, few of these papers dissected the potential effects of classical drug treatments of patients with neurodegenerative diseases as an important confounding factor that can induce a distinct signature, as previously shown in such diseases (figure 3).117
Indeed, a similar comparison can be made with some seminal studies published a couple of years ago, which showed differences in the compositions of the gut microbiota of type 2 diabetic and non-diabetic patients including an increase in A. muciniphila.118 After a couple of years, it was finally discovered that the previous data (at least on this bacterium) were in fact flawed because of the drug treatments (ie, metformin, liraglutide) (figure 3).102 103 118 119 A similar observation is done for the use of proton pump inhibitor that strongly affect the gut microbiota.120–122
Thus, one may argue that we should take into account of such confounding factor for all the future studies, including neurodegenerative disorders.
Indeed, in such diseases, the vast majority of patients experience changes in dietary habits, chronic constipation and other GI comorbidities; therefore, these pathologies are classically associated with a change in gut motility and other premotor symptoms. Interestingly, it has been well described that stool frequency and consistency (figure 3)123 are predominant factors for intestinal A. muciniphila abundance. In fact, a recent paper highlights the crucial importance of assessing stool frequency and consistency in microbiome studies.123 In addition, caloric restriction and fasting periods are key characteristics of the feeding behaviour of patients with neurodegenerative diseases, and these factors have clearly been shown to increase the abundance of A. muciniphila in the human gut.124
Therefore, these different examples strongly show us that we need a careful analysis and demonstration of causality before making claims about the risks or beneficial effects of some microbes. As stated above, the last decade has been characterised by an abundance of papers assessing the difference between the microbiomes of healthy or diseased cohorts. Hence, the misinterpretation of data and claims that a bacterium is involved in a phenotype based on changes in the abundance of the bacterium is still seen almost daily in the literature (figure 3).
Finally, in addition to the case of P. copri and A. muciniphila, recent data combining both observational and proof-of-concept studies have also suggested that specific species might be directly involved in the protection against metabolic disorders associated with obesity (eg, Christensenella minuta)125 or, conversely, might contribute to the onset of such metabolic disorders (eg, Ralstonia pickettii, Enterobacter cloacae).126 127 Thus, these kinds of findings are interesting but cannot be generalised and warrant further confirmation using extensive studies, as detailed previously.
Conclusion and perspectives
We are living with a tremendous amount of microorganisms in our guts, ranging from bacteria, archaea to virus and fungi. There is no doubt about the fact that we have progressed in the analysis of the composition of the microbiota and of the key metabolites produced and even in the discovery and isolation of novel bacteria. However, we must acknowledge that a large number of the studies published the last couple of years have reported differences in the microbiome under different conditions. Although most of them are very well performed, we still need more work to go beyond the simple associations and we need to provide as much as possible more complex analysis (eg, multiomics and time series measurements) if we want to finally approach the final causality. Indeed, simple associations may lead to misinterpretation or overselling of expected results when translated into the human context. Thus, both types of approaches are important, that is, comparing diseased and healthy conditions and then showing the causality as a proof of concept. However, the general population as well as health practitioners should be rigorous when drawing conclusions from papers that assume that the discovery of differences in gut microbiota composition is potentially strongly associated with a specific disease or its overall evolution (box 1).
Key definitions
Microbiota: This term refers to a collection of all taxa constituting microbial communities, such as bacteria, archeae, fungi and protists. When it refers to a specific environment, the term is preceded by the said location, for example, ‘the gut microbiota’ refers to the intestinal tract and the ‘oral microbiota’ is used when speaking about all the microbes from the oral cavity.128
Microbiome: This term was initially used to refer to the genes harboured by microbes; however, currently, the term ‘microbiome’ is also commonly used to refer to the microorganisms themselves (ie, the microbiota).128
Probiotics: These are live microorganisms that, when administered in adequate amounts, confer a health benefit to the host.129
Prebiotics: These are substrates that are selectively utilised by host microorganisms conferring health benefits.130
Metagenome: This term refers to the entire genetic material present in a sample. The metagenome is composed of the genomes of several individual organisms, for instance, the genomes of human cells and of the gut microorganisms that are present in faecal material.
Metabolome: This term refers to the quantitative complement of all the low molecular-weight molecules present in a biological sample.131
Virome: This term refers to the entire viral genetic material present in a sample. The virome consists of the genomes of viruses.
Acknowledgments
I profoundly acknowledge the tremendous work and effort of my research team and I am deeply grateful for their constant support. PDC is a senior research associate at FRS-FNRS (Fonds de la Recherche Scientifique).
References
Footnotes
Contributors PDC wrote the review.
Funding PDC is a recipient of grants from FNRS, FRFS-WELBIO, under grant WELBIO-CR-2017-C02. This research was supported by the FRS-FNRS under The Excellence Of Science (EOS 30770923). This work is supported in part by the Funds Baillet Latour (Grant for Medical Research 2015). PDC is a recipient of the POC ERC grant 2016 (European Research Council, Microbes4U_713547) and ERC Starting Grant 2013 (Starting grant 336452-ENIGMO).
Competing interests PDC is inventor on patent applications dealing with the use of A. muciniphila and its components in the treatment of obesity and related disorders. PDC is co-founder of A-Mansia biotech SA.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.