Article Text

Abstract
Objective There is a striking association between human cholestatic liver disease (CLD) and inflammatory bowel disease. However, the functional implications for intestinal microbiota and inflammasome-mediated innate immune response in CLD remain elusive. Here we investigated the functional role of gut–liver crosstalk for CLD in the murine Mdr2 knockout (Mdr2−/−) model resembling human primary sclerosing cholangitis (PSC).
Design Male Mdr2 −/−, Mdr2−/− crossed with hepatocyte-specific deletion of caspase-8 (Mdr2−/− /Casp8∆hepa) and wild-type (WT) control mice were housed for 8 or 52 weeks, respectively, to characterise the impact of Mdr2 deletion on liver and gut including bile acid and microbiota profiling. To block caspase activation, a pan-caspase inhibitor (IDN-7314) was administered. Finally, the functional role of Mdr2−/− -associated intestinal dysbiosis was studied by microbiota transfer experiments.
Results Mdr2−/− mice displayed an unfavourable intestinal microbiota signature and pronounced NLRP3 inflammasome activation within the gut–liver axis. Intestinal dysbiosis in Mdr2−/− mice prompted intestinal barrier dysfunction and increased bacterial translocation amplifying the hepatic NLRP3-mediated innate immune response. Transfer of Mdr2−/− microbiota into healthy WT control mice induced significant liver injury in recipient mice, highlighting the causal role of intestinal dysbiosis for disease progression. Strikingly, IDN-7314 dampened inflammasome activation, ameliorated liver injury, reversed serum bile acid profile and cholestasis-associated microbiota signature.
Conclusions MDR2-associated cholestasis triggers intestinal dysbiosis. In turn, translocation of endotoxin into the portal vein and subsequent NLRP3 inflammasome activation contribute to higher liver injury. This process does not essentially depend on caspase-8 in hepatocytes, but can be blocked by IDN-7314.
- primary sclerosing cholangitis
- cholestatic liver diseases
- gut inflammation
- enteric bacterial microflora
- intestinal barrier function
Statistics from Altmetric.com
- primary sclerosing cholangitis
- cholestatic liver diseases
- gut inflammation
- enteric bacterial microflora
- intestinal barrier function
Significance of this study
What is already known on this subject?
Primary sclerosing cholangitis (PSC) exhibits remarkable associations with inflammatory bowel disease (IBD) and enteric microbial dysbiosis.
The liver plays a central role at the intersection between the host and the gut commensal microbiota.
Gut microbial dysbiosis can trigger disruption of the intestinal barrier leading to bacterial translocation, which consecutively activates immune cells to release various proinflammatory cytokines and chemokines in the liver.
The colonic mucosa is an important site for NLRP3 activation, which is involved in the regulation of intestinal homeostasis.
Significance of this study
What are the new findings?
Activation of Caspase-1 in gut and liver of Mdr2−/− mice plays an essential role in disease progression in the Mdr2 model.
Caspase-8 in hepatocytes is dispensable for progression of cholestatic liver disease (CLD) in Mdr2−/− mice.
Increased bacterial translocation and NLRP3 inflammasome activation within the gut–liver axis is involved in the pathogenesis of CLD in Mdr2−/− mice and is associated with an altered enterohepatic circulation of bile acids and gut microbiota composition.
Transfer of microbiota derived from Mdr2−/− mice into healthy WT control mice induces significant intestinal dysbiosis and liver injury in WT mice indicating a causal role of Mdr2−/− microbiota in liver disease progression.
The pan-caspase inhibitor IDN-7314 blocks NLRP3 activation within the gut–liver axis, partially reverses changes in microbiota composition and results in a marked improvement of CLD.
How might it impact on clinical practice in the foreseeable future?
The genetic defect of Mdr2 triggers intestinal dysbiosis, which contributes to disease progression. Owing to its high plasticity, gut microbiota modulation may represent a therapeutic target in humans with CLD.
NLRP3 activation plays pivotal roles in mediating Mdr2−/− -induced gut–liver interaction. Hence, NLPR3 inflammasome may be relevant to human PSC.
IDN-7314 blocks disease progression in the Mdr2−/− mouse model and thus could be a therapeutic option for PSC.
Introduction
Primary sclerosing cholangitis (PSC) is an idiopathic, chronic, cholestatic liver disease (CLD) characterised by chronic biliary inflammation, periductal fibrosis and biliary tree destruction. PSC frequently progresses to liver cirrhosis and has a significant risk for developing cholangiocarcinoma and gall bladder carcinoma.1 Genetic, immunological and environmental factors contribute to disease progression. However, the aetiology and pathogenesis of PSC remain poorly understood.
Genetic deletion of multidrug resistance protein 2 (Mdr2−/− ) in mice causes a complete absence of phosphatidylcholine in bile. This change leads to increased biliary concentrations of non-micellar-bound, free bile acids, resulting in sclerosing cholangitis and cholelithiasis associated with progressive liver injury.2 The Mdr2−/− model closely resembles the course of PSC in humans and represents an appropriate PSC animal model3 to study relevant PSC-dependent mechanisms of disease progression.
Inflammasomes are crucial mediators of the innate immune response that are activated on recognition of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). Among the nucleotide-binding oligomerisation domain-like receptor inflammasome complexes, the NLRP3 (also known as NALP3) inflammasome has most widely been characterised and is an important signalling node.4–7 After sensing of PAMPs or DAMPs, activated NLRP3 serves as a scaffold to recruit the adapter apoptosis-associated speck-like protein (ASC) containing a C-terminal caspase recruitment domain, resulting in the activation of pro-caspase-1 into its cleaved form.8 Activated caspase-1 cleaves the precursor cytokines, precursor interleukin-1 beta (pro-IL-1β) and precursor interleukin-18 (pro-IL-18), respectively, generating the bioactive cytokines IL-1β and IL-18. Furthermore, caspase-1 is also able to induce an inflammatory form of cell death known as pyroptosis.5
NLRP3 activation is implicated in the pathogenesis of many liver diseases such as ischaemia–reperfusion injury, drug-mediated, pathogen-mediated or endotoxin-mediated liver damage, as well as alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD).9 Recent findings showed that NLRP3 contributes to the maintenance of intestinal homeostasis and modulation of gut microbiota, which in turn influence the intestine and distant organs.6 The colonic mucosa is an important site for NLRP3 activation, where it is implicated in regulating intestinal homeostasis. The role of NLRP3 for experimental colitis is a matter of controversy and needs further clarification, while data on its role in experimental PSC models remain scarce.
PSC exhibits remarkable associations with inflammatory bowel disease (IBD) and enteric microbial dysbiosis.10 The liver is located at the intersection between the host and the gut commensal microbiota.11 Gut microbial dysbiosis can trigger disruption of the intestinal barrier leading to bacterial translocation, which consecutively activates immune cells to release various proinflammatory cytokines and chemokines in the liver.12 Bacterial translocation has been increasingly recognised as a crucial factor in the pathogenesis of ALD and NAFLD.13 14 Here, bile acids are directly involved in modulating intestinal microbiota composition by direct and indirect antimicrobial effects.15
Conversely, gut microbiota metabolises bile acids in the intestine and thus alters signalling via bile acid receptors.16 Several clinical studies have observed changes in intestinal microbiota signature in patients with PSC.17–20 Here, different microbiota composition and reduced species diversity were evident in patients with PSC compared with healthy controls. As bile acids shape intestinal microbiota composition and are at the same time subject to microbial enzymatic modifications, it seems not far to seek that the change in bile acid composition may impact microbiota composition. However, due to the obvious experimental restrictions of these clinical studies, the functional role of intestinal microbiota for PSC pathogenesis has not been addressed experimentally. Hence, the causal role of microbiota for gut–liver crosstalk in PSC pathogenesis remains largely unknown.
In our present study, we aimed to close this gap by dissecting the functional role of gut microbiota and gut–liver crosstalk in PSC pathogenesis using the preclinical Mdr2−/− mouse model. Our present work identifies gut microbiota as a driver of CLD and highlights the NLRP3 inflammasome as an important interface mediating inflammation within the gut–liver axis.
Materials and methods
Mice
Mdr2−/− and wild-type (WT) control mice of the FVB/N background and male gender were housed in the Central Animal Facility University Hospital RWTH Aachen. All mice analysed at the 52 weeks time point were littermates of the same heterozygous breeding pair. Mice for the 8 weeks time point were generated with two homozygous breeding pairs, which were derived from the initial heterozygous breeding pair (F1 generation). All mice were housed in the same specific pathogen-free room in individually ventilated cages with free access to a standard chow diet and water. Mice with deletion of caspase-8 in hepatocytes and biliary cells (Casp8∆hepa) have been described earlier.21 We crossed Mdr2−/− mice with Casp8∆hepa animals to generate Mdr2−/− /Casp8∆hepa double-knockout animals and used Cre-negative floxed (Mdr2−/− /Casp8f/f) littermates as control (C57Bl/6 background). All the mice were treated in accordance to the criteria of the German and Spanish administrative panels on laboratory animal care and approved by the local Animal Care Committees (#AZ84-02.04.2013.[CT], #AZ84-02.04.2017.A327 [CT] and PROEX 195/16 [FJC], PROEX 210/18 [FJC], respectively).
Patients
The analysed patient samples were randomly selected from the human biobank at the University Hospital Magdeburg. The ethical review board of the Medical Faculty at the University of Magdeburg approved the study (‘Erforschung und Charakterisierung von NLRP3 und assoziierten Proteinen in Cholestase-assoziierten Erkrankungen’, 61/18) based on the international (ICH,GCP) and national guidelines. All patients gave consent. The clinical investigation has been conducted according to the principles expressed in the 1975 Declaration of Helsinki. Histological assessment and scoring for inflammasome activation were performed by a board-certified liver pathologist in a blinded fashion.
Methods
Bile acids were analysed by ultra-performance liquid chromatography-tandem mass spectrometry (Wallenberg Laboratory at Sahlgrenska University Hospital, Sweden). Western blot, qRT-PCR, caspase activity assay, immunostaining, histological analysis, microbiota data analysis, flow cytometry analysis of intrahepatic leucocytes, endotoxin measurement, intestinal permeability test and statistical analysis are described in detail in supporting information available online (see the online supplementary material).
Treatment with a pan-caspase inhibitor (IDN-7314)
Pan-caspase inhibitor IDN-7314 (Conatus, California, 0.3 mg/mL dilution with 0.5% carboxymethylcellulose sodium suspension) was administered to 4-week-old Mdr2−/− and WT mice on a daily basis by oral gavage in a dose of 3 mg/kg. Treatment was performed for 28 days. IDN-7314 has been tested to inhibit the activity of caspase-1 (IC50=0.222 nM), −3 (IC50=5.38 nM), −8 (IC50=0.138 nM) and −9 (IC50=2.08 nM).
Results
Mdr2 deficiency results in cholestatic liver injury
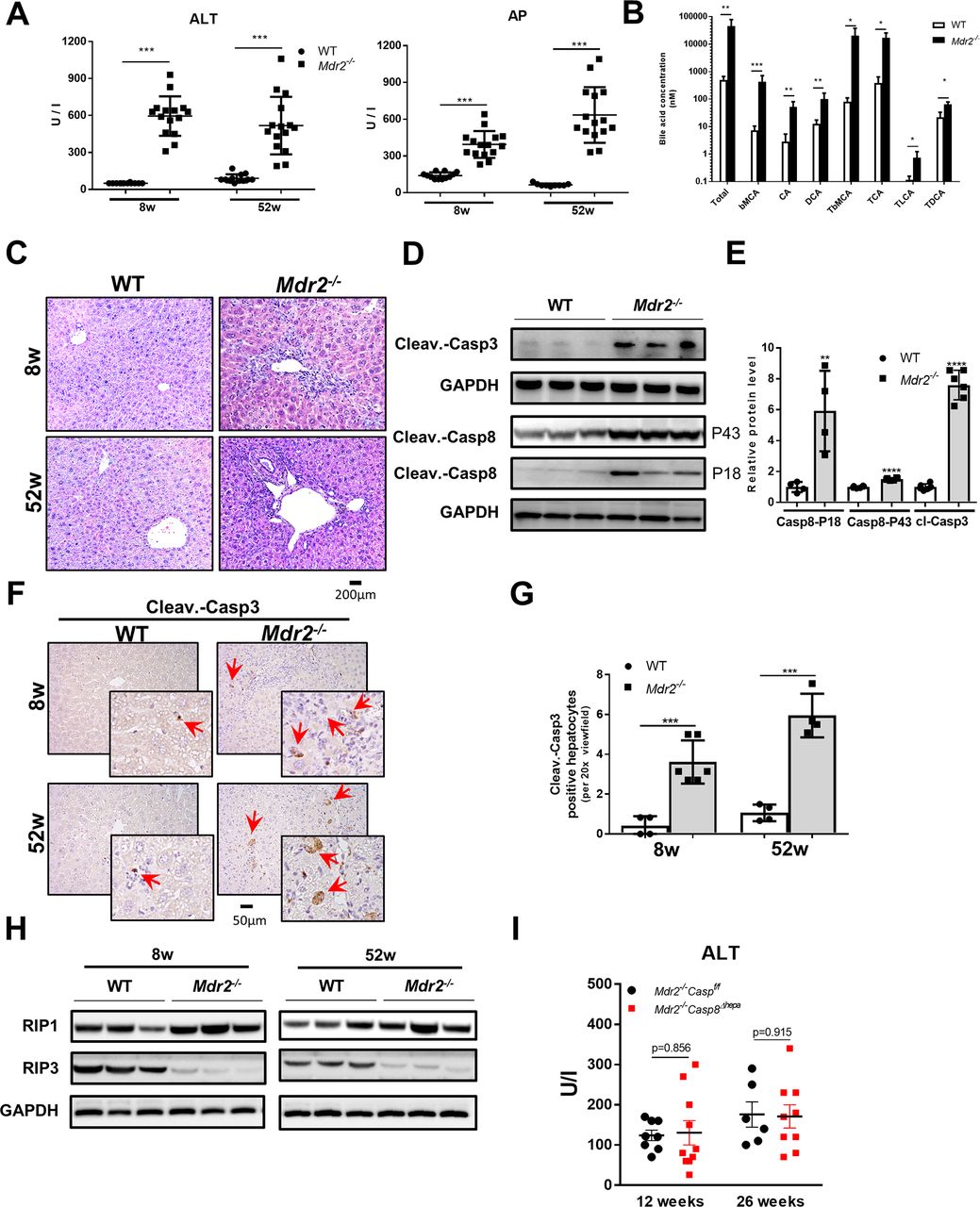
Mdr2−/− mice showed significantly increased serum aminotransferase levels at 8 weeks and 52 weeks (figure 1A, left panel and see the online supplementary fig ure 1A), revealing serious liver damage. Consistent with previous reports,22 23 Mdr2−/− mice showed a higher liver versus body weight ratio compared with WT mice (see the online supplementary figure 1B). Furthermore, the biliary system in Mdr2−/− mice showed obstruction associated with profoundly increased serum concentrations of alkaline phosphatase (AP, figure 1A, right panel) and bile acids (figure 1B) compared with WT mice. Synthesis of bile acids is the major pathway of cholesterol catabolism in mammals; in Mdr2−/− mice, lower serum cholesterol levels were evident (see the online supplementary figure 1C).
Supplemental material
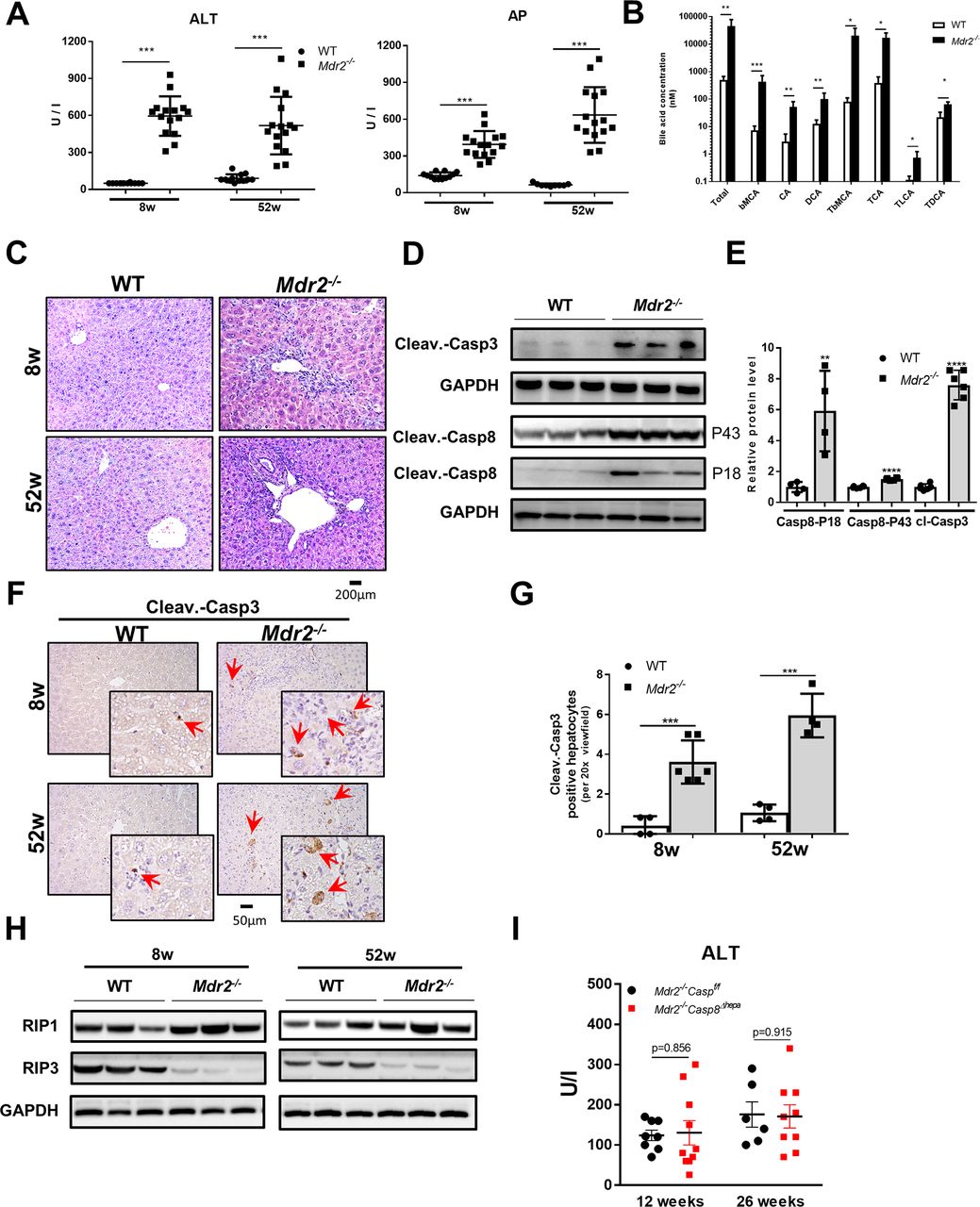
Mdr2 deficiency results in cholestatic liver injury and triggers apoptotic cell death. (A) Serum ALT and AP levels of 8-week-old and 52-week-old Mdr2−/− mice and respective littermate controls (WT) (mean ±SD, n=12–15). (B) Serum bile acid concentrations of 8-week-old Mdr2−/− and WT mice were determined (mean ±SD, n=6). (C) Representative liver sections after H&E staining. (D) Protein levels of activated caspase-3 and caspase-8 (P18 and P43) were determined by western blot analyses in livers of Mdr2−/− and WT mice at 8 weeks of age. (E) Protein level quantification of caspase-3 and caspase-8 western blots (mean ±SD, n=3–6). (F) Immunohistochemical staining against cleaved (ie, activated) caspase-3 was performed in livers of Mdr2−/− and WT mice at 8 and 52 weeks of age. Red arrows depict cells with caspase-3 activation (left panel). (G) The number of cells with activated caspase-3 were determined for each mouse group and time point (mean ±SD, n=6–8). Protein levels of (H) RIP1 and RIP3 (mean ±SD, n=6–8) were determined by western blot analysis in livers of Mdr2−/− and WT mice at 8 and 52 weeks of age. (I) Serum ALT levels were determined from Mdr2−/− /Casp8f/f and Mdr2−/− /Casp8∆hepa mice at the age of 12 and 26 weeks (mean ±SD, n=6–9). GAPDH was used as loading control in western blot analysis. Data are considered significant if *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AP, alkaline phosphatase; bMCA, β-muricholate acid; CA, cholic acid; cleav.-casp3, cleaved caspase-3; cleav.- casp8, cleaved caspase-8; DCA, deoxycholic acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TbMCA, tauro-β-muricholate acid; TCA, taurocholate acid; TDCA, taurodeoxycholate acid; TLCA, taurolithocholic acid; WT, wild type.
Liver histology of Mdr2−/− mice presented a strong chronic periductular inflammatory response, progressive bile duct proliferation and periportal fibrosis developing over time (52 weeks) (figure 1C and see the online supplementary figure 1D and E). Consequently, all Mdr2−/− male mice presented liver tumours macroscopically after 1 year (see the online supplementary figure 1F). Liver tumours were analysed histologically by a board-certified liver pathologist and classified as well-differentiated hepatocellular tumours with focal atypia and small group necrosis (see the online supplementary figure 1G). Livers of Mdr2−/− mice presented a strong ductular reaction with portal inflammation and apoptosis (see the online supplementary figure 1H and I). This elevated apoptosis rate and the inflammatory response was followed by an increased mitotic activity. Immunohistochemical (IHC) staining against Ki-67 confirmed increased cell cycle activity in 52-week-old Mdr2−/− mice compared with WT littermate controls (see the online supplementary figure 2A).
Supplemental material

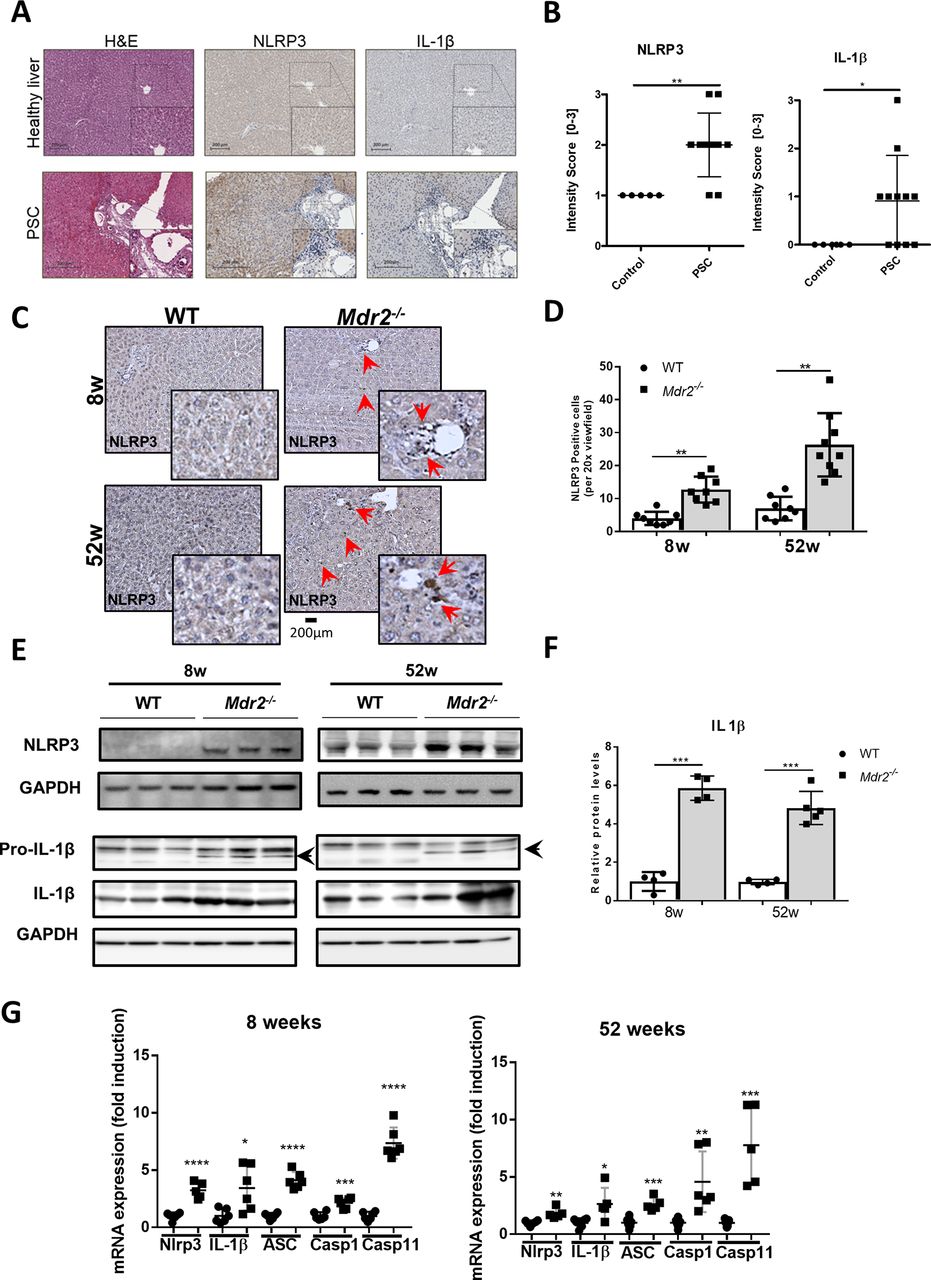
NLRP3 inflammasome is activated in human PSC and Mdr2−/− livers. Livers of Mdr2−/− and WT mice at 8 and 52 weeks of age were included in this analysis. (A) Livers from patients with PSC (n=11) show increased NLRP3 and IL-1β-positive cells around the periportal tract compared with healthy controls (n=5). (B) NLRP3 and IL-1β intensity scoring of IHC stainings performed by a board-certified liver pathologist. (C) Representative NLRP3 immunohistochemical staining on paraffin liver sections are shown. Red arrows indicate NLRP3-positive cells. (D) NLRP3-positive cells were quantified for each mouse group and time point. (E) NLRP3, pro-IL-1β and IL-1β protein levels were determined by western blot analysis and (F) quantified. GAPDH was used as loading control. (G) mRNA expressions of NLRP3, IL-1β, ASC, caspase-1 and caspase-11 were determined by qRT-PCR in livers. Data are expressed as the mean ±SD from 6 to 12 mice per group and considered significant if *p<0.05, **p<0.01, ***p<0.001, **** p < 0.0001 . ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL-1β, interleukin-1 beta; PSC, primary sclerosing cholangitis; pro-IL-1β, precursor interleukin-1 beta; WT, wild type.
Immune cell infiltration is a central pathogenic feature of acute and chronic liver injury. Fluorescence-activated cell sorting (FACS) analysis and IHC staining against CD45 revealed significantly increased leucocyte infiltration in Mdr2−/− livers compared with WT controls (see the online supplementary figure 2B). Mdr2−/− livers showed significantly increased MCP-1 expression (see the online supplementary figure 2C) which was associated with infiltration of inflammatory monocyte-derived macrophages (MoMFs) (defined as F4/80low CD11bhi) as evidenced by FACS (see the online supplementary figure 2D). Inflammatory cell infiltration was confirmed by F4/80 IHC staining showing pronounced immune cell infiltration in Mdr2−/− livers, especially around the periportal area (see the online supplementary figure 2E), consistent with higher CD68 and F4/80 liver mRNA expression (see the online supplementary figure 2F).
Altogether, these data showed increased inflammation in Mdr2−/− livers characterised by recruitment of MoMFs, thereby stimulating progression of a PSC-like phenotype.
Caspase-8-dependent apoptosis signalling is dispensable for disease progression in Mdr2−/− mice
Accumulation of bile acids, especially those with hydrophobic properties, triggers cholestasis-induced hepatocyte cell death.24 Mdr2−/− livers displayed significantly higher cleaved caspase-3 and caspase-8 expressions (figure 1D and E). Additionally, caspase-3 and caspase-8 enzyme activities were significantly increased in Mdr2−/− compared with WT livers (see the online supplementary figure 3A). Furthermore, the number of cells with activated caspase-3 was significantly enhanced in Mdr2−/− livers, and these cells were mostly located around bile ducts (figure 1F and G). Moreover, caspase-9 protein expression was higher in Mdr2−/− compared with WT livers (see the online supplementary figure 3B).
Supplemental material

Gut microbiota composition is altered in Mdr2−/− mice. Caecal content of 52-week-old Mdr2−/− and WT littermate mice was included in this analysis. (A) NMDS graph based on Bray-Curtis dissimilarity shows clear separation of Mdr2−/− and WT mice. (B) Bar chart based on permutational multivariate analysis of variance (ADONIS) presenting the percentage of variability of the gut microbiota explained by the factors ‘genotype’, ‘litter’ and ‘cage’. (C) Microbiota alpha diversity in Mdr2−/− and WT mice presented as species richness. (D) Computational linear discriminant analysis (LDA)effect size (LEfSe) analysis identifies the taxa Lachnospiraceae to be differentially regulated in Mdr2−/− (red) and WT (blue) mice. Data expressed as the mean ±SD from 11 to 12 mice per group and considered significant if *p<0.05, **p<0.01, ***p<0.001. LDA, ; NMDS, non-metric multidimensional scaling; WT, wild type.
Receptor-interacting protein kinase 3 (RIP3/RIPK3) has emerged as a critical regulator of programmed necroptosis.25 Hence, we measured RIP1 and RIP3 protein expression in Mdr2−/− livers. RIP3 protein expression was abrogated, while RIP1 was slightly upregulated in Mdr2−/− compared with WT livers (figure 1H and see the online supplementary figure 3C). In addition, mRNA expressions of other markers such as Bcl2 and Bax involved in cell apoptosis were significantly increased in Mdr2−/− livers (see the online supplementary figure 3D). Accordingly, these results suggest that liver injury in Mdr2−/− mice was likely mediated by apoptosis but not through necroptosis.
After we found that caspase-8 is strongly activated in Mdr2−/− livers (figure 1D and E), we next aimed to understand its role for disease progression. We crossed Mdr2−/− animals with Casp8Δhepa mice to generate Mdr2−/− /Casp8∆hepa animals. Importantly, male Mdr2−/− /Casp8∆hepa mice did not reveal significant differences in transaminase levels at the age of 12 and 26 weeks when compared with male Mdr2−/− animals (figure 1I). Additionally, no significant difference in liver versus body weight ratio was found between Mdr2−/− /Casp8Δhepa and Mdr2−/− animals (see the online supplementary figure 4A). Moreover, fibrosis progression between Mdr2−/− /Casp8∆hepa and Mdr2−/− livers was not significantly different (see the online supplementary figure 4B). Thus, these results suggest that caspase-8 in hepatocytes is dispensable for disease progression in Mdr2−/− livers despite its strong activation.
Supplemental material

Mdr2 deletion leads to intestinal barrier disruption. Colon and ileum of 8-week-old and 52-week-old Mdr2−/− and WT mice were included in the analyses (A–D). (A) ZO-1 protein expression was determined by western blot analysis of colon and ileum tissue specimens. GAPDH was used as loading control. (B) ZO-1 mRNA expression was determined by qRT-PCR in colon and ileum samples. (C) Representative ZO-1 immunofluorescence staining was performed on frozen colon and ileum sections. (D) Representative immunofluorescence staining against mucin-2 was performed on Carnoy-fixed colon sections. (E) Intestinal permeability to FITC-dextran was measured in 8-week-old Mdr2−/− and WT mice. (F) Portal vein LPS concentration was measured in Mdr2−/− and WT mice at 8 weeks of age. (G) The relative abundance of bacterial 16S ribosomal RNA genes of total bacteria (left panel), Enterobacteriaceae and Enterococcus (right panel) was determined by qPCR using DNA isolated from liver tissue. Data were normalised to the total amount of genomic DNA and GAPDH. Data are expressed as the mean ±SD from 6 to 11 mice per group/time point and considered significant if *p<0.05, **p<0.01, ***p<0.001. FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LPS, lipopolysaccharide; WT, wild type; ZO-1, zonula occludens-1.
Pronounced activation of the NLRP3 inflammasome in human PSC and Mdr2−/− livers
Inflammasomes are intracellular signalling platforms activating caspase-1, which then cleaves pro-IL-1β into active IL-1β.26 The NLRP3 inflammasome has been implicated in various liver diseases. To investigate the relevance of NLRP3 activation for PSC, human liver sections of patients with PSC and healthy control patients were stained against NLRP3 (figure 2A). Interestingly, IHC of patients with PSC showed a significant increase in NLRP3 and IL1-β-positive cells; IL1-β-positive cells indicated inflammasome activation, which was absent in healthy controls (figure 2A and B). We examined the correlation of the grade of liver fibrosis with the NLRP3-, IL-1β and clinical MELD score in patient samples. The degree of NLRP3 and IL-1β activation were increased in more advanced cases (r=0.6528, p=0.0039 for NLRP3; r=0.8083, p=0.0007 for IL-1β; see the online supplementary figure 4C). Furthermore, the MELD score calculated from clinical parameters correlated with the grade of fibrosis (r=0.8322, p=0.0005; see the online supplementary figure 4C and table 1). Based on these findings, we investigated the role of NLRP3 in the context of inflammasome activation in Mdr2−/− mice. IHC in 8-week-old and 52-week-old Mdr2−/− livers showed a strong increase of NLRP3-positive cells (figure 2C and D). Additionally, protein overexpressions of NLRP3, pro-IL-1β and IL-1β in Mdr2−/− livers were found (figure 2E and F and see the online supplementary figure 4D), which were consistent with higher mRNA levels for NLRP3, IL-1β, ASC, caspase-1 and caspase-11 in these liver samples (figure 2G). These results demonstrate that cholestatic liver injury in Mdr2−/− mice is mediated by pronounced inflammasome activation.
Supplementary file 9
Alteration in gut microbiota composition of Mdr2−/− mice
In Mdr2−/− mice, we found a major change in serum bile acid composition. As changes in bile acid metabolism may impact enterohepatic circulation and thus alter microbiota composition, we investigated whether Mdr2−/− mice show intestinal dysbiosis. While PCoA axis 1 and 2 of weighted UniFrac Distances did not show a clear clustering, non-metric multidimensional scaling (NMDS) analysis based on Bray-Curtis dissimilarity displayed clear separation between 52-week-old Mdr2−/− and WT littermate mice (figure 3A and see the online supplementary figure 4H); litter and cage information were included in these analyses and highlighted in colour (see the online supplementary figure 4G). To evaluate the relative contribution of Mdr2 deficiency on the observed differences between groups, we performed permutational multivariate analysis of variance (ADONIS)27 based on the Bray-Curtis dissimilarity matrix considering the factors ‘genotype’, ‘litter’ and ‘cage’. As expected, individual litter and cage both had a significant impact on microbiota community structure. Cage explained a significant and large proportion of up to 34% (R2 0.34, ***p<0.0006) of total microbiota variability based on permutational multivariate analysis of variance (ADONIS). The individual litter also had a significant impact on microbiota signature and explained up to 0.25% (R2 0.25, **p<0.0012). Importantly, despite the observed cage effect, these independent co-housing experiments revealed a reproducible effect of the Mdr2−/− genotype on microbiota composition, which also explained a significant proportion of up to 10% of the gut microbiota variability (R2 0.098, **p<0.006) (figure 3B).
Gut microbiota composition in Mdr2−/− and WT mice was mainly dominated by bacterial members belonging to Clostridiales (family Lachnospiraceae) and in minor proportion of Bacteroidales (see the online supplementary figure 4E). Reduced microbial species diversity was observed in Mdr2−/− compared with WT mice (figure 3C). By performing computational LDA effect size (LEfSe) analysis28 on all genera, bacteria of the group of Lachnospiraceae stood out as they were most prominently regulated between WT and Mdr2−/− littermates (figure 3D). While several bacterial operational taxonomic units (OTUs) belonging to the group of Lachnospiraceae were increased in WT mice, Lachnospiraceae-uncultured_OTU_69 and Lachnospiraceae-OTU_2 were differentially increased in Mdr2−/− mice (figure 3D). Based on 16 s-V4 sequence similarity, the OTU_69 and OTU_2 belong to the family Lachnospiraceae. However, the Silva database v128 did not support deeper classification of this OTU. Interestingly, Lachnospiraceae have been implicated in human PSC and are characterised as producing iso-bile acids species, suggesting that the observed changes in bile acid composition may influence microbial bile acid metabolism.19 29 Importantly, we observed a significant correlation between Lachnospiraceae-uncultured_OTU_69 abundance and serum ALT levels pointing to a pathophysiological role during CLD (see the online supplementary figure 4F).
In sum, these data indicate that CLD in Mdr2−/− mice results in a markedly altered gut microbiota composition and leads to a specific microbiota signature.
Deletion of Mdr2 leads to intestinal barrier disruption
Gut dysbiosis may induce changes in intestinal barrier function. Therefore, we investigated the gut epithelium. Histological analyses of H&E-stained tissue sections did not reveal significant differences between Mdr2−/− and WT mice (see the online supplementary figure 5A and B).
Supplemental material
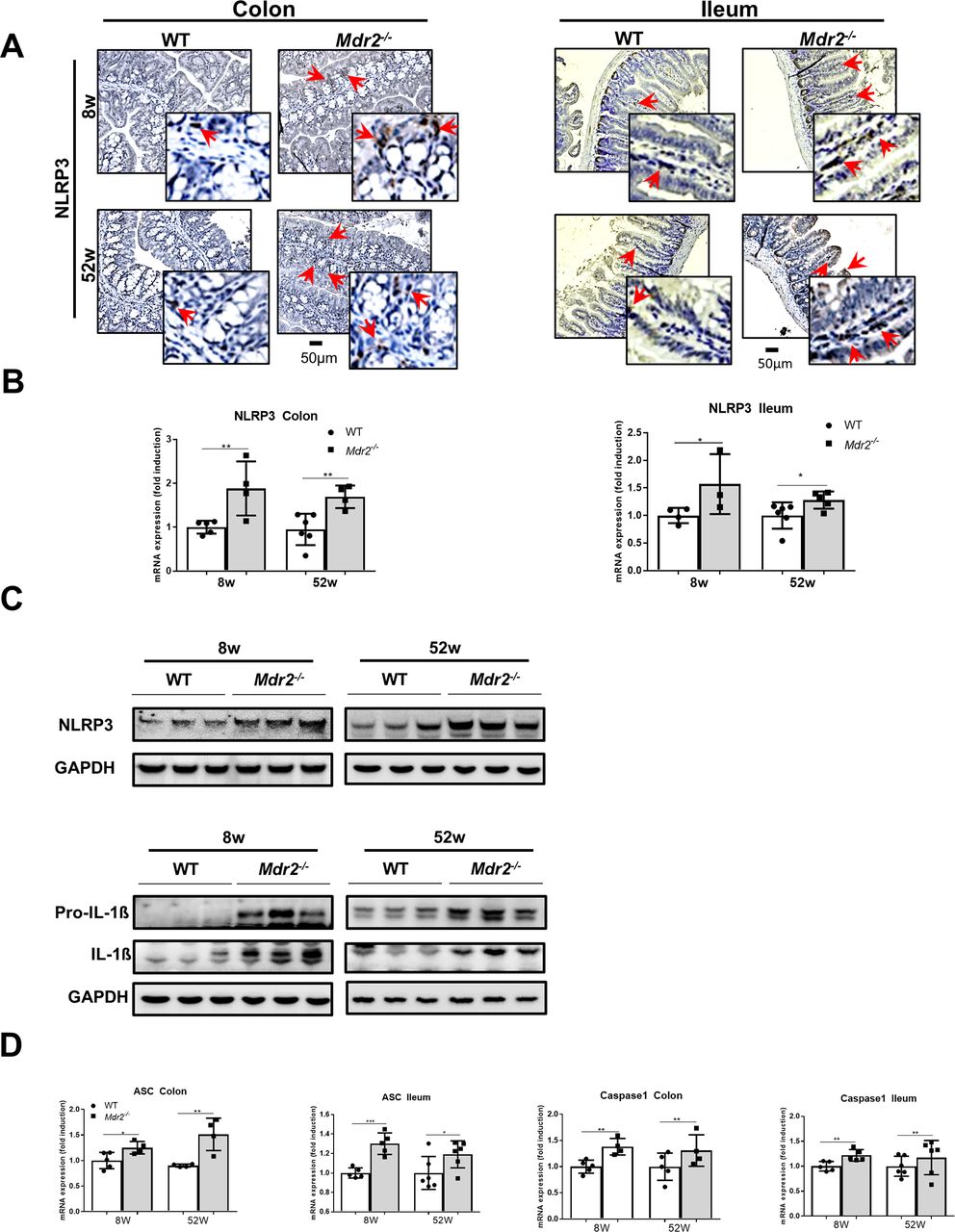
The NLRP3 inflammasome is activated in the intestine of Mdr2−/− mice. Intestines of 8-week-old and 52-week-old Mdr2−/− and WT mice were included in this analysis. (A) NLRP3 immunohistochemical staining was performed on paraffin colon and ileum sections. Red arrows indicate NLRP3-positive cells. (B) NLRP3 mRNA expression was determined by qRT-PCR in colon and ileum. (C) NLRP3, pro-IL-1β and IL-1β western blot analyses were performed in the colon. GAPDH was used as loading control. (D) ASC and caspase-1 mRNA expressions were determined by qRT-PCR in colon and ileum. Data are expressed as the mean ±SD from six to eight mice per group and considered significant if *p<0.05, **p<0.01, ***p<0.001. ASC, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain; GADPH, glyceraldehyde-3-phosphate dehydrogenase; pro-IL-1β, precursor interleukin-1 beta; WT, wild type.
Epithelial tight junctions maintain intestinal barrier where zonula occludens-1 (ZO-1), a scaffolding protein, plays a pivotal role in tight junction formation. ZO-1 protein and mRNA expression were significantly decreased in the colon and ileum of Mdr2−/− mice at different time points (online supplementary figure 4A and B and see the online supplementary figure 5C). Immunostaining further confirmed ZO-1 downregulation in the gut villus epithelium at apical and basolateral cell regions of Mdr2−/− mice (figure 4C). Furthermore, in the colon of Mdr2−/− mice, integrity of the mucus layers was disturbed and mucus was significantly thinner in Mdr2−/− compared with WT mice (figure 4D, online supplementary figure 5D).
To investigate whether reduced tight junction expression and mucus layers translated into increased permeability, Mdr2−/− and WT mice were orally gavaged with fluorescein isothiocyanate conjugated dextran (FITC-dextran). While WT mice displayed very low serum levels of FITC-dextran 4 hours after administration, spectrophotofluorometry showed significantly higher FITC-dextran concentrations in serum of Mdr2−/− mice (figure 4E). Increased permeability was further confirmed by measuring lipopolysaccharide (LPS) concentrations in portal venous blood (figure 4F). Mdr2−/− mice displayed significantly higher LPS levels in portal blood compared with WT controls, while LPS levels in central venous blood were not different between the groups.
Finally, to assess bacterial translocation in livers of Mdr2−/− and WT mice, the relative abundance of bacterial 16S ribosomal RNA genes normalised to the total amount of genomic DNA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was determined by qPCR using DNA isolated from liver tissue. Strikingly, livers of Mdr2−/− mice showed a significantly increased relative abundance of rRNA genes of total bacteria and Enterobacteriaceae (normalised to GAPDH) compared with WT littermate controls (figure 4G). Furthermore, Enterococcus rRNA genes were detectable in all Mdr2−/− livers at the 52-week time point, while it was undetectable in all WT control mice (figure 4G, right panel). Altogether, these data show impaired barrier function in Mdr2−/− mice prompting increased intestinal permeability and translocation of microbe-associated molecular patterns (MAMPs).
Mdr2 deletion activates inflammasome signalling in the gut
The healthy gut has a robust and balanced immune system. On the one hand, it prevents detrimental collateral damage induced by inflammatory responses against the commensal microbiota and antigens, on the other hand, it ensures protection against invading pathogens. Increased infiltration of CD45+immune cells was found in the intestine of Mdr2−/− mice (see the online supplementary figure 6A). Additionally, there were more CD11b and F4/80-positive cells in the colon and ileum of Mdr2−/− mice (see the online supplementary figure 6B and C). This was associated with higher F4/80 mRNA levels in Mdr2−/− mice (see the online supplementary figure 6D), triggering increased proinflammatory cytokine expression of, for example, tumour necrosis factor-alpha (see the online supplementary figure 6E).
Supplemental material

Transfer of Mdr2−/− microbiota induces intestinal dysbiosis in healthy WT mice. (A) WT and Mdr2−/− microbiota was transferred into WT littermate mice via oral gavage every second day for 1 week. (B) ALT and AST serum levels were measured. (C) Western blot analysis was performed to determine NLRP3, pro-IL-1β, IL-1β and caspase-1 protein expression in colons and livers of WTFMT(WT) and WTFMT(Mdr2−/−) mice. (D and E) Expression of tight junction protein ZO-1 was quantified by western blot in the ileum of WT mice receiving WT microbiota (WTFMT(WT)) and mice receiving Mdr2−/− microbiota (WTFMT(Mdr2−/−)). (F) FITC-dextran serum concentrations measured 4 hours after oral gavage of FITC-dextran in WTFMT(WT) and WTFMT(Mdr2−/−) mice. (G) FACS data showing relative abundance of MoMFs (defined as Ly6G-, CD11bhi, F4/80+) in livers. (H) DESeq analysis comparing microbiota community structure of WTFMT(Mdr2−/−) mice before the first transfer of Mdr2−/− microbiota (day 0) to microbiota after transfer (day 7). (I) Microbiota community structure of WTFMT(WT) mice before the first transfer of WT microbiota (day 0) to microbiota after transfer (day 7). Data are expressed as the mean ±SD from four to eight mice per group and considered significant if *p<0.05, **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate transaminase; MoMF, monocyte-derived macrophages; FACS, fluorescence-activated cell sorting; FITC-dextran, fluorescein isothiocyanate conjugated dextran; pro-IL-1β, precursor interleukin-1 beta; WT, wild type; ZO-1, zonula occludens-1.
As we already observed increased inflammasome activation in Mdr2−/− livers, we wondered if NLRP3 activation might also be involved in mediating the gut phenotype in these animals. Eight-week-old and 52-week-old Mdr2−/− mice exhibited substantially higher NLRP3-positive cells in paraffin-embedded colon and ileum sections as evidenced by immunohistochemistry compared with WT mice (figure 5A). These findings go along with higher NLRP3 mRNA expression in the colon and ileum of Mdr2−/− mice (figure 5B). Additionally, increased NLRP3, pro-IL-1β and IL-1β protein expressions were found in the colon of Mdr2−/− mice (figure 5C and see the online supplementary figure 6F), and RT-qPCR analysis demonstrated strong ASC and caspase-1 upregulation (figure 5D). These data suggest that intestinal NLRP3 inflammasome activation in Mdr2−/− mice is associated with liver disease progression.
Transfer of Mdr2−/− microbiota induces intestinal dysbiosis in healthy WT mice
As changes in gut microbiota composition of Mdr2−/− mice were associated with increased NLRP3 inflammasome activation and loss of intestinal barrier integrity, we next sought to investigate the effect of the Mdr2−/− microbiota on intestinal homeostasis. To this end, microbiota derived from Mdr2−/− or WT control mice was transferred via oral gavage into healthy WT mice for 1 week (online supplementary figure 6A). Strikingly, faecal microbiota transfer (FMT) of Mdr2−/− , but not WT-derived microbiota resulted in a significant increase of serum transaminase activity pointing to elevated liver injury in recipient WT mice (online supplementary figures 6B and 7A). This phenotype was mediated by a pronounced NLRP3 inflammasome activation in the colon and liver of recipient WTFMT(Mdr2−/−) mice (figure 6C). FMT of Mdr2−/− -derived microbiota suppressed ZO-1 tight junction expression in WTFMT(Mdr2−/−) mice as evidenced by western blot analyses, immunofluorescence staining and increased gut permeability in the FITC-dextran assay (figure 6D-F, see the online supplementary figure 7B). IHC staining against CD45 showed increased leucocyte infiltration in WTFMT(Mdr2−/−) mice compared with WTFMT(WT) littermates (see the online supplementary figure 7C). The inflammatory infiltrates were dominated by MoMFs (defined as Lg6G-, CD11bhi, F4/80+), suggesting that these cells are mediators of the observed phenotype and early sensors of gut-derived inflammatory signals.
Supplemental material

IDN-7314 treatment ameliorates liver injury in Mdr2−/− mice. Four-week-old Mdr2−/− and WT mice were treated by oral gavage daily with 3 mg/kg IDN-7314 or vehicle alone for 4 weeks. (A) Liver injury was determined by analysing biochemical serum parameters (AST, ALT, AP and GLDH). (B) Representative H&E-stained liver paraffin sections are shown. (C) Total bile acids, beta-muricholic acid (β-MCA), deoxycholic acid (DCA) and tauro beta-muricholic acid (TbMCA) concentrations were analysed in serum. (D) Representative immunohistochemical staining of cleaved caspase-8 (left panel) and quantification (right panel) on liver paraffin sections are depicted. (E) Cleaved caspase-8 and cleaved caspase-3 protein levels were determined by western blot analysis of IDN-7314 or vehicle-treated Mdr2−/− and WT livers. GAPDH was used as loading control. (F) Caspase-3 enzyme activity was analysed in livers of WT and Mdr2−/− mice with and without IDN-7314 treatment. Data are expressed as the mean±SD from 8 to 11 mice per group and considered significant if **p<0.01, ***p<0.001. ALT, alanine aminotransferase; AST, aspartate transaminase; AP, alkaline phosphatase; cleav.-casp3, cleaved caspase-3; cleav.-casp3, cleaved caspase-8; GADPH, glyceraldehyde-3-phosphate dehydrogenase; GLDH, glutamate dehydrogenase; WT, wild type.
We observed strong differences in microbiota community structure of WT and Mdr2−/− donor mice (online supplementary figure 7D and E). Differences were found in Lachnospiraceae, which were also identified in LEfSe analysis of 52-week-old WT and Mdr2−/− littermate mice (see the online supplementary figure 7E and figure 3D). Interestingly, transfer of Mdr2−/− -derived microbiota induced significant changes in the bacterial community structure of recipient WT mice (figure 6H). When comparing microbiota composition of WTFMT(Mdr2−/−) before (day 0) and after (day 7) FMT, differential gene expression analysis based on the negative binomial distribution (DESeq230) analysis revealed in total 12 bacterial groups that were significantly upregulated and 8 that were downregulated on microbiota transfer (figure 6H). Interestingly, three differentially regulated OTUs were part of the family Lachnospiraceae and we observed an expansion of Bacteroidales S24-7 group in WTFMT(Mdr2−/−) mice, which may have the colitogenic potential.31 In contrast, on transfer of WT microbiota, only minor changes were observed in three groups all belonging to the family Lactobacillus (figure 6I). After FMT, microbiota composition of WTFMT(Mdr2−/−) was similar to Mdr2−/− donor microbiota, while microbiota of WTFMT(Mdr2−/−) differed in several OTUs from WTFMT(WT) littermate microbiota (see the online supplementary figure 7F and G).
Collectively, these data demonstrate that intestinal dysbiosis, as observed in Mdr2−/− mice, is transmissible via FMT and contributes to inflammatory liver disease.
Pan-caspase inhibitor IDN-7314 treatment ameliorates liver injury in Mdr2−/− mice
In Mdr2−/− livers, we found activation of distinct caspases. While caspase-8 deletion in hepatocytes did not rescue the phenotype, we found a strong activation of caspase-1 and the NLRP3 inflammasome. Therefore, we sought to test systemic pan-caspase inhibition—especially caspase-1 and caspase-8—by treating animals with IDN-7314. Eight-week-old Mdr2−/− mice treated with IDN-7314 displayed significantly reduced liver injury as evidenced by respective serum parameters (figure 7A). Additionally, H&E staining demonstrated decreased periportal inflammation and lower bile duct proliferation in IDN-7314-treated Mdr2−/− livers compared with vehicle-treated mice (figure 7B).
These changes were associated with significantly decreased serum bile acid concentrations in Mdr2−/− mice after IDN-7314 treatment (figure 7C). To test the functional effect of IDN-7314 on caspase inhibition, we investigated the caspase-8 activity. IHC staining and immunoblot analyses showed strongly decreased caspase-8 expression in Mdr2−/− livers after IDN-7314 treatment (figure 7D and E and see the online supplementary figure 8A). Furthermore, cleaved caspase-3 protein expression (figure 7E and see the online supplementary figure 8B) and caspase-3 enzyme activity (figure 7F) were significantly decreased in Mdr2−/− livers after IDN-7314 treatment.
Supplemental material

![[gutjnl-2018-316670supp001.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp002.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp003.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp004.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp006.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC6/embed/inline-supplementary-material-6.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp007.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC7/embed/inline-supplementary-material-7.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp008.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC8/embed/inline-supplementary-material-8.jpg?download=true){kind=link}
{kind=link}
![[gutjnl-2018-316670supp009.jpg]](https://gut.bmj.com/content/gutjnl/68/8/1477/DC9/embed/inline-supplementary-material-9.jpg?download=true){kind=link}
{kind=link}
IDN-7314 treatment inhibits inflammasome activation in Mdr2−/− mice. (A) Lachnospiraceae OTU_2 is significantly reduced after IDN-7314 treatment of Mdr2−/− mice compared with vehicle treatment. (B) Principal coordinate analysis (NMDS) graph based on Bray-Curtis dissimilarity shows separation of Mdr2−/− littermate mice with and without IDN-7314 treatment. Ellipses have been computed in R using the multivariate t-distribution at 0.95 confidence level. (C) Western blot analysis and (D) quantification of ZO-1 protein expression in the colon of IDN-7314 or vehicle-treated mice. (E) Western blot analyses of IDN-7314 or vehicle-treated Mdr2−/− and WT livers and (F) colons were performed to determine NLRP3, pro-IL-1β, IL-1β and caspase-1 expression. GAPDH was used as loading control. (G) Hepatic mRNA expression of NLRP3, IL-1β, ASC, caspase-1, F4/80 and IL-6 were determined by qRT-PCR in IDN-7314 or vehicle-treated Mdr2−/− and WT livers. Data are expressed as the mean±SD from six mice per group and considered significant if *p<0.05, **p<0.01, ***p<0.001. ASC, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain; GADPH, glyceraldehyde-3-phosphate dehydrogenase; NMDS, non-metric multidimensional scaling; OTU, operational taxonomic unit; pro-IL-β, precursor interleukin-1 beta; ZO-1, zonula occludens-1.
IDN-7314 treatment reshapes microbiota composition and inhibits inflammasome activation in Mdr2−/− livers
Strikingly, IDN-7314 treatment did not only significantly ameliorate liver disease and reduce serum bile acid concentrations, but also partially reversed the Mdr2−/− microbiota signature (figure 8A and B and see the online supplementary figure 8C). Of note, >20% of total gut microbiota variability in Mdr2−/− littermate mice±IDN-7314 (R2 0.2303, p<0.0077**, ADONIS) could be explained by the factor ‘IDN-7314 treatment’ (see the online supplementary figure 8C). We next analysed the impact of pan-caspase inhibitor treatment on bacteria that separated 52-week-old Mdr2−/− and WT mice in our LEfSe analysis (figure 3D). Lachnospiraceae (OTU2), initially overexpanded in Mdr2−/− compared with WT microbiota, were now significantly reduced after IDN-7314 treatment (figure 8A). Together these data highlight that caspase inhibition can modulate gut–liver crosstalk by reshaping the unfavourable microbiota signature as found in Mdr2−/− mice. Strikingly, caspase inhibition via IDN-7314 improved intestinal barrier integrity in Mdr2−/− mice as evidenced by ZO-1 western blot (figure 8C and D).
As intestinal dysbiosis can drive hepatic inflammasome activation and our results demonstrated strong inflammasome expression with disease progression within the gut–liver axis of Mdr2−/− mice, we investigated the impact of IDN-7314 treatment on inflammasome activation. Western blot analysis showed decreased protein expression of NLRP3, pro-IL-1β, IL-1β and caspase-1 in colon and liver of Mdr2−/− mice after IDN-7314 treatment (figure 8E and F and see the online supplementary figure 8D). IDN-7314 administration resulted in significantly reduced mRNA expression of NLRP3, ASC, IL-1β and caspase-1 in Mdr2−/− livers (figure 8G). Concomitant with these findings, mRNA expression of hepatic F4/80 and IL-6 were significantly decreased in Mdr2−/− livers after IDN-7314 treatment compared with vehicle-treated Mdr2−/− mice (figure 8G). As MoMFs were identified as important mediators of the observed inflammatory response in Mdr2−/− mice, we performed immunofluorescence staining against CD11b to analyse the effect of IDN-7314 treatment on immune cell infiltration. In line with H&E histology, we observed a significantly reduced number of CD11b+ cells in IDN-7314-treated Mdr2−/− mice compared with vehicle-treated Mdr2−/− controls (see the online supplementary figure 8E).
In summary, these results show that caspase activation significantly contributes to disease progression in Mdr2−/− mice and suggest that NLRP3 inflammasome activation plays a prominent role in this process.
Discussion
The Mdr2−/− mouse is considered to recapitulate human PSC. In this model, loss of the phospholipid transporter Mdr2, also known as Abcb4, triggers a cholestatic response, which induces intrahepatic sclerosing cholangitis characterised by biliary fibrosis. Here, we investigated the pathways contributing to liver inflammation and injury in order to define new molecular targets, which might also be relevant for human PSC.
The relationship between gut and liver in PSC—in virtue of its striking association with chronic IBD—is known for decades. However, until now, pathogenetic connections linking IBD to PSC remain scarce.32 About 80% of patients with PSC suffer from IBD (especially ulcerative colitis but also Crohn’s disease) and up to 8% of patients with IBD also have PSC.33 Interestingly, patients with IBD and PSC frequently have more progressive liver disease. Conversely, patients who have undergone liver transplantation due to PSC often suffer from more severe IBD.34 Interestingly, colectomy prior to PSC diagnosis is associated with decreased risk for liver transplantation or death.35 While positive family history and male gender are well-established risk factors for PSC,36 accumulating evidence points to a role of gut microbiota in disease pathogenesis.37 In PSC, several cross-sectional studies demonstrated a reduced gut alpha diversity and significant alterations in the overall microbiota composition compared with healthy controls.17–19 While the link between changes in microbiota composition and PSC has been established, until now, the biological relevance and pathogenetic role of microbiota for gut–liver crosstalk remain incompletely understood.37
In our present study, we show for the first time that the intestinal microbiota of Mdr2−/− mice differs significantly from WT littermate controls. Mdr2−/− microbiota showed reduced species diversity and significant alterations in the family of Lachnospiraceae, which have the important ability to form secondary bile acids by multistep 7α-dehydroxylation. Similarly, patients with PSC display intestinal dysbiosis, independent of potentially accompanying chronic IBD.19 Earlier studies could not dissect whether changes in microbiota composition were secondary to liver disease or actually played a direct pathogenetic role. In our current study, we aimed to characterise these findings. Concomitant with our previous data,38 intestinal dysbiosis in Mdr2−/− mice was linked to altered mucus layers, reduced expression of tight junction proteins and increased bacterial translocation. These data indicate that changes in bile acid composition and flow in Mdr2−/− mice significantly affect the intestine and impair intestinal barrier integrity.
To characterise the mechanism underlying intestinal dysbiosis in Mdr2−/− mice, we analysed intestinal immune cell infiltrates histologically. The intestinal lamina propria is particularly enriched in leucocytes, which actually outnumber epithelial cells.39 These leucocytes protect against infection, control host-microbiota mutualism and maintain barrier integrity.40 41 F4/80+gut resident macrophages are strategically located in the intestinal lamina propria and serve important functions in maintaining gut-barrier integrity.41 Interestingly, disruption of the epithelial barrier in Mdr2−/− mice was followed by pronounced macrophage infiltration, which exhibited high NLRP3 expression. The NLRP3 inflammasome is a multiprotein complex that orchestrates innate immune responses to infection and cell damage and is an important sensor at the host–microbial interface.42 43 Bacterial products can represent both the first and second signal to activate the NLRP3 inflammasome.42 On activation, the inflammasome complex cleaves pro-IL-1β into its active form, which exerts pro-inflammatory functions. Indeed, in concert with induced gene expression of inflammasome-related genes, western blot analyses of ileum and colon specimen revealed elevated levels of active IL-1β protein in Mdr2−/− mice. Together, our current data show increased activation of the NLRP3 inflammasome in dysbiotic Mdr2−/− mice. Here, increased protein levels of active IL-1β are associated with disruption of intestinal barrier integrity.
Bacterial products can induce NLRP3 inflammasome activation, but also bile acids shape NLRP3 inflammasome activity.44–46 Accumulating evidence highlight bile acids as pleiotropic signalling molecules with manifold activities far beyond their function as detergents of dietary lipids.47 48 Importantly, bile acids serve as mediators between host metabolism and inflammation bridging gut and liver within the enterohepatic circulation.47 Recent data identify the primary bile acid CDCA, which was also found to be strongly increased in Mdr2−/− mice, as an inducer of NLRP3.46 In contrast, in LPS-primed bone marrow-derived macrophages treated with primary or secondary bile acids, NLRP3 inflammasome activation is inhibited via the G-protein-coupled bile acid receptor, Gpbar1 - cyclic adenosine monophosphate - protein kinase A (TGR5-cAMP-PKA) axis.44 Hence, different bile acid species might have opposing effects on the NLRP3-mediated immune response depending on their target of the action. Bile acids also influence gut microbiota composition due to their intrinsic antimicrobial properties.16 Conversely, changes in gut microbiota composition affect microbial bile acid metabolism and thus shape bile acid-mediated biological activities.47 In our current study, we are unable to dissect, whether increased intestinal NLRP3 activation is mediated by changes in luminal bile acid content or a consequence of altered gut microbiota in Mdr2−/− mice. However, we clearly demonstrate that NLRP3 activation is an important feature of the observed phenotype. Accordingly, inhibition of NLRP3 activity via IDN-7314 partially reversed changes in microbiota composition and improved intestinal barrier integrity in Mdr2−/− mice.
In recent years, the reciprocal interplay between gut and liver, also termed gut–liver axis, during health and disease gained increasing attention and emerged as a field of intense research activities. Loss of intestinal barrier integrity and disturbed microbiota composition, as also observed in Mdr2−/− mice, aggravate chronic liver diseases ranging from NAFLD,38 ALD49 as well as liver fibrosis50 and hepatocellular carcinoma.51
In our study, increased gut permeability prompted bacterial translocation as evidenced by increased LPS levels in the portal vein and bacterial DNA in livers of Mdr2−/− mice. As reported earlier in the context of non-alcoholic steatohepatitis (NASH), translocation of MAMPs triggers an NLRP3-mediated inflammatory response and infiltration of MoMFs. Recent data from Maroni et al demonstrate that expression of the NLRP3 inflammasome is increased in reactive cholangiocytes in human PSC as well as the murine DDC model of PSC. The presented in vitro data link NLRP3 activation to epithelial barrier dysfunction of cholangiocytes, which might be involved in the development of experimental biliary injury and contribute to the progression of PSC in humans. In an earlier study, Jahnel et al used an elegant experimental approach to study the impact of intestinal inflammation on the liver.52 In their study, chronic colitis induced by dextran sulfate sodium treatment triggered mild portal inflammation in otherwise healthy heterozygous Mdr2 mice.52 Collectively, these data suggested that intestinal dysbiosis drives liver disease in Mdr2−/− mice.
To finally prove that intestinal dysbiosis of Mdr2−/− mice alone triggers liver disease progression, we performed FMT experiments. Healthy WT mice transplanted with Mdr2−/− microbiota partially recapitulated intestinal dysbiosis as evident in Mdr2−/− mice, displayed infiltration of inflammatory macrophages in the liver and increased liver function tests (LFTs). Together, our data show for the first time that CLD in Mdr2−/− mice induces pathogenic changes in microbiota composition and loss of intestinal homeostasis. As also reported for NASH,53 intestinal microbiota triggers a hepatic innate immune response. This phenotype was transmissible via FMT and contributed to the progression of liver disease. Hence, these data highlight the causal role of gut microbiota in CLD. Although microbiota analyses confirmed successful FMT, we still cannot completely exclude that the observed phenotype might also be driven by metabolites that were transferred. To exclude this bias, microbiota would have to be cultured ex vivo before transfer and separated from intestinal metabolites. These culture experiments and potential further targeted isolation studies were beyond the scope of our current study, but a fascinating path for future investigations.
In the context of NASH, microbiota depletion with broad-spectrum antibiotics could ameliorate liver disease, and germ-free mice are protected from diet-induced obesity.38 54 Contrasting to these data, intestinal microbiota has been shown to protect against CCL4 and MCD diet-induced liver fibrosis.55 56 Similarly, data from Tabibian et al demonstrate that germ-free Mdr2−/− mice actually develop more severe liver disease, which might support a protective function of gut microbiota in CLD.23 Germ-free mice completely lack microbial bile acid metabolism and consequently, their bile acid pool consists entirely of primary bile acids, including ursodeoxycholic acid, >95% conjugated with taurine. Of note, despite 2.5-times larger bile acid pool, germ-free mice do not show a predisposition to CLD.57 Hence, these data argue for a differential role of microbiota in the Mdr2−/− model. While the complete absence of gut microbiota may aggravate the Mdr2−/− phenotype, dysbiotic microbiota induce NLRP3-dependent inflammation and thus fuel disease progression.
In Mdr2−/− mice, intestinal dysbiosis and translocation of MAMPs were accompanied by activation of different caspases, including caspase-8 and caspase-1 in the gut and liver of Mdr2−/− animals. To dissect the pathogenetic role of caspases in this model, we introduced Casp8∆hepa mice and pharmacologically blocked caspase activation using the pan-caspase inhibitor IDN-7314. At the used treatment doses, the pan-caspase inhibitor IDN-7314 primarily blocks caspase-1 and caspase-8. While hepatocyte-specific inhibition of caspase-8 could not attenuate liver injury in Mdr2−/− mice, IDN-7314 blocked disease progression and decreased liver injury by attenuating caspase-1 and inflammasome activation. Therefore, these data suggest that in the Mdr2−/− model, NLRP3 activation in gut and liver perpetuates liver injury and thus contributes to disease progression beyond the primary genetic defect in the MDR2 transport system.
In summary, our results demonstrate that gut microbiota and intestinal dysbiosis are important drivers of CLD in Mdr2−/− mice. Importantly, our data point to a direct pathogenetic role of cholestasis-induced intestinal dysbiosis for liver disease progression. Here, the NLRP3 inflammasome is a core mediator within the gut–liver axis. Gut microbiota and NLRP3 inflammasome represent interesting therapeutic targets for CLD, which might also be relevant in humans.
Acknowledgments
The authors would like to thank Thomas Ritz for helpful discussions and Conatus Pharmaceuticals for providing IDN-7314.
References
Footnotes
LL and KMS contributed equally.
FJC and CT contributed equally.
Contributors LL and KMS: performed most of the experiments, analysed most of the data and drafted the manuscript. MF and HS: contributed to the performance of experiments. EJCG and TS: analysed the microbiota composition. H-UM and AW: performed bile acids analyses. AM: managed the experimental mice. JP: provided technical support. JH: analysed double knock-out mice. JR: contributed to the FITC-dextran detection. MH and HWZ: provided critical revision of the manuscript. TL, JH and PP: analysed liver histology. IB and AN: measured and analysed LPS levels. CL: provided the double knock-out mice data, data interpretation and critically reviewed the manuscript. FJC: supervised and reviewed the study. CT: designed the experiments, supervised the study, drafted the paper and provided funds.
Funding This study was supported by the German Research Foundation TR 285/10‑1 and SFB/TRR 57 to CT; the Federal Ministry of Education and Research (ObiHep grant #01KU1214A to CT); the Liver-LiSyM grant (BMBF) to CT, The HDHL-INTIMIC Di-Mi-Liv to CT and KMS; the SFB 985 project C3 to CT; the Interdisciplinary Centre for Clinical Research (START Grant #691438) within the Faculty of Medicine at RWTH Aachen University, the Deutsche Krebshilfe (project #109988 to CT and CL); the Interdisciplinary Centre for Clinical Research within the faculty of Medicine at the RWTH Aachen University (IZKF E8-2 to CL); the Helmholtz Association (VH-NG-933 to TS); the START-Program of the Faculty of Medicine #691405, RWTH Aachen), i³tm Seed Fund Project SF_15_5_17 (RWTH Aachen), the MINECO Retos SAF2016-78711, EXOHEP-CM S2017/BMD-3727, NanoLiver-CM Y2018/NMT-4949, ERAB Ref. EA 18/14, AMMF 2018/117 and COST Action CA17112 and Ramón y Cajal Fellowship (RYC-2014-15242) to FJC; The Swedish Research Council and the Regional Research Council of Västra Götaland to H-UM; Pudong New Area Health and Family Planning Commission Clinical peak discipline construction (Shanghai, PWYgf2018-05), Natural Science Foundation of Jiangxi Province (S2019ZRMSB2159) to LL
Competing interests None declared.
Ethics approval Approval by the local Animal Care Committees (#AZ84-02.04.2013.A184(CT), #AZ84-02.04.2017.A327(CT), PROEX 195/16 (FJC) and PROEX 210/18 (FJC), respectively.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.