Article Text

Abstract
Background and aims Leucocyte migration to gut mucosa, mediated by integrin binding to mucosal addressin cell adhesion molecule (MAdCAM), is a promising target for therapeutic intervention in inflammatory bowel disease. This first-in-human study of a monoclonal antibody to MAdCAM, PF-00547,659, aimed to explore the safety and preliminary efficacy of this gut-specific mechanism in ulcerative colitis.
Methods In this randomised, double-blind placebo-controlled study, 80 patients with active ulcerative colitis received single or multiple (three doses, 4-week intervals) doses of PF-00547,659 0.03–10 mg/kg IV/SC, or placebo. Safety was assessed by adverse events, laboratory tests, and immunogenicity. Exploratory efficacy analyses were based on Mayo score and endoscopic responder rates at weeks 4 and 12. Faecal calprotectin was quantified as a measure of disease activity, and the number of α4β7+ lymphocytes was measured to demonstrate drug activity.
Results No obvious drug-related side effects were observed in the PF-00547,659 group, while patient numbers, especially those fully exposed, were small. Overall responder/remission rates at 4 and 12 weeks were 52%/13% and 42%/22%, respectively with combined PF-00547,659 doses compared with 32%/11% and 21%/0%, respectively with placebo. Equivalent endoscopic responder rates were 50% and 42% versus 26% and 29%, respectively. Faecal calprotectin levels decreased to a greater extent with PF-00547,659 than placebo (week 4: 63% vs 18%). Despite variability, there was a trend for an increase in α4β7+ lymphocytes in patients receiving PF-00547,659.
Conclusions The favourable short-term safety profile and preliminary efficacy findings for PF-00547,659 in this first-in-human study pave the way for further investigation in larger trials, to establish the role of PF-00547,659 in ulcerative colitis treatment.
Trial Register No: NCT00928681.
- MAdCAM
- ulcerative colitis
- mucosal vascular addressin cell adhesion molecule 1 protein
- human monoclonal antibody
- endothelium
- inflammatory bowel disease
- adhesion molecules
- IBD
- IBD clinical
Statistics from Altmetric.com
- MAdCAM
- ulcerative colitis
- mucosal vascular addressin cell adhesion molecule 1 protein
- human monoclonal antibody
- endothelium
- inflammatory bowel disease
- adhesion molecules
- IBD
- IBD clinical
Significance of this study
What is already known about this subject?
Despite the fact that biological therapies targeting tumour necrosis factor α (TNFα) have significantly advanced the management of patients with inflammatory bowel disease (IBD), there is a remaining need for efficacious drugs with good safety profiles.
Homing of leucocytes to the gut mucosa is a promising target for therapeutic intervention in IBD, shown by studies with the α4-integrin antibody natalizumab in Crohn's disease and ulcerative colitis.
However, natalizumab is not available to treat Crohn's disease in Europe given the risk of progressive multifocal leucoencephalopathy (PML).
To avoid this, more selective strategies targeting the α4β7/MAdCAM pathway in the gut would be expected to confer similar efficacy as seen with natalizumab but a better safety profile.
What are the new findings?
This is the first study of PF-00547,659, a highly specific, fully human anti-MAdCAM IgG2 antibody in patients with active ulcerative colitis.
Study results show that single and multiple doses of PF-00547,659 were safe and well tolerated with no evidence of immunogenicity.
Furthermore, preliminary efficacy findings were promising with higher response and remission rates for PF-00547,659 as compared to placebo.
How might it impact on clinical practice in the foreseeable future?
PF-00547,659 is a promising target for larger phase II studies in ulcerative colitis.
Introduction
Biological therapies have an established role in the management of moderate-to-severe inflammatory bowel disease (IBD) that is refractory to conventional treatments. Currently, the only biological treatments for IBD which are licensed worldwide target tumour necrosis factor α (TNFα) (infliximab, adalimumab and certolizumab pegol for Crohn's disease, and infliximab for ulcerative colitis). While anti-TNF agents provide a significant advance in the treatment of refractory IBD, with short-term clinical response rates of approximately 60–70% in ulcerative colitis and Crohn's disease, primary and secondary treatment failure remain a problem for many patients.1–4 Furthermore, there are safety concerns with anti-TNF agents, including susceptibility to intracellular opportunistic infections such as tuberculosis, and the potential risk of lymphoma and other malignancies.5 6 Therefore, a need remains for efficacious drugs with good safety profiles, to improve patient outcomes in the debilitating conditions of Crohn's disease and ulcerative colitis.
Homing of leucocytes to the gut mucosa is a promising target for therapeutic intervention in IBD, shown by studies with the α4-integrin antibody natalizumab in Crohn's disease7–9 and ulcerative colitis.10 However, the use of natalizumab is limited by well-publicised concerns over the risk of progressive multifocal leucoencephalopathy (PML),11–13 an opportunistic infection of the central nervous system (CNS) by the polyomavirus JC virus.14 Susceptibility to PML may arise as a result of disruption of immune surveillance in the CNS, which may be mediated in part by homing of α4β1-positive leucocytes to vascular cell adhesion molecule (VCAM).15–18 The effects of natalizumab in the gut likely involve—among other mechanisms (including inhibition of α4β1 integrin)—another population of cells expressing α4-integrin–α4β7-positive (α4β7+) leucocytes, which bind mucosal addressin cell adhesion molecule (MAdCAM).19–21
MAdCAM is expressed predominantly on vascular endothelium in the intestinal lamina propria.22 Animal studies confirm the importance of MAdCAM in gut inflammation in colitis,23–25 and suggest that VCAM has a comparatively minor role.26–28 Selectively targeting the α4β7/MAdCAM pathway, to attenuate leucocyte trafficking in the gut without disrupting VCAM-mediated leucocyte homing in other organs, would be expected to confer similar efficacy in IBD to that seen with a less selective anti-leucocyte trafficking mechanism, but with potential safety benefits due to the more localised effect. The concept of selectively targeting the α4β7/MAdCAM pathway for IBD treatment is supported by animal studies,23 29 and initial clinical results with vedolizumab (MLN0002), a humanised monoclonal antibody for α4β7-integrin.30 31
While natalizumab and vedolizumab target integrins, blockade of MAdCAM itself is a gut-specific therapeutic mechanism for IBD. Importantly, for reducing the risk of CNS infections such as PML, immunohistochemistry studies confirm an absence of MAdCAM expression in the human brain.32
PF-00547,659 is a highly specific, fully human anti-MAdCAM IgG2 antibody.33 We report the first study of PF-00547,659 in humans, in patients with active ulcerative colitis (NCT00928681, http://ClinicalTrials.gov/). The primary objective was to investigate the safety and tolerability of single and multiple doses of PF-00547,659. We also performed exploratory efficacy assessments based on clinical and endoscopic indices of ulcerative colitis activity, and explored effects on number of circulating CD4+ α4β7+ lymphocytes (a mechanistic biomarker), faecal calprotectin (a biomarker for disease activity), and plasma high-sensitivity C reactive protein (hsCRP; a biomarker for inflammation).
Methods
Study design, patients and setting
This was a randomised, double-blind (sponsor-open), placebo-controlled, dose-escalating, parallel-group study. Patients were assigned to one of six single-dose or five multiple-dose cohorts, and within each cohort were randomised to receive placebo or PF-00547,659. Randomisation was conducted using a sequential numbering system based on the order of patient enrolment.
In the single-dose study phase, patients received single intravenous (IV) infusion of PF-00547,659 at doses of 0.03, 0.1, 0.3, 1.0 or 10 mg/kg or a single subcutaneous (SC) injection of PF-00547,659 at dose of 3.0 mg/kg, or matching placebo. Patients in the multiple-dose phase received three doses, 4 weeks apart, of PF-00547,659 at doses of 0.1, 0.3 or 3.0 mg/kg IV or 0.3 or 1.0 mg/kg SC, or placebo (figure 1). PF-00547659 was presented as a sterile, isotonic solution containing 10 mg/ml PF-00547659 in an acetate buffer solution containing mannitol, Polysorbate and ethylenediaminetetraacetic acid (pH 5.5). Sodium chloride solution 0.9% w/v BP was used as the placebo.

Study flow diagram. Patients who discontinued the study due to lack of efficacy or adverse events were considered treatment failures and were included as non-responders in efficacy analyses. AE, adverse event; IV, intravenous; SC, subcutaneous; UC, ulcerative colitis.
Follow-up continued for 12 weeks in the single-dose phase and 16 weeks in the multiple-dose phase. If PF-00547,659 remained detectable in blood plasma at the last clinic visit, follow-up continued at 4-week intervals until drug concentrations fell below the detection limit of 20 ng/ml. To maintain blinding, some patients on placebo were followed up over an extended period.
Inclusion criteria
Male and female patients aged 18–70 years, with a histologically confirmed diagnosis of ulcerative colitis for at least 3 months prior to study entry, were enrolled. Patients were required to have active ulcerative colitis (total Mayo score ≥6, endoscopic subscore ≥2), despite being on stable doses of 5-aminosalicylic acid (5-ASA) or sulfasalazine for 3 weeks; or azathioprine or 6-mercaptopurine for 3 months, which were to be continued throughout the study; or oral steroids (up to 40 mg/day prednisolone or equivalent) for 2 weeks, which could be tapered at the investigator's discretion.
Exclusion criteria
Patients were excluded from the study if they had ulcerative colitis confined to proctitis; fulminant colitis; history of malignant neoplasia; renal or hepatic impairment; significant cardiovascular disease in the previous 12 months; hepatitis B, hepatitis C or HIV infection; evidence of pathogenic infection in faecal culture; history of steroid dependence (determined at the investigator's discretion); or allergic disease or sensitivity to heparin.
Patients were also excluded if they had undergone surgery for ulcerative colitis or were likely to require surgery during the study, or if they had used methotrexate, mycophenolate, cyclosporine or biological therapy within 3 months prior to study dosing, interferon or leucocyte apheresis within 12 months, intravenous steroids within 1 month, any rectally administered treatment within 1 week, or had previously used natalizumab or PF-00547,659. Women were excluded if they were pregnant or lactating.
Ethics
The research protocol was approved by the appropriate institutional review board or independent ethics committee for each study site. All participants gave written informed consent.
Study procedures
Safety evaluations
All adverse events (AEs) and the likely relationship to study treatment were recorded. Additional safety evaluations included laboratory tests, vital signs, and 12-lead ECG. To assess whether PF-00547,659 induced an immunogenic response, antidrug antibodies were measured in serum from blood samples taken predose and every 4 weeks up to week 12.
In the evaluation of efficacy and biomarker data, week 4 of the single-dose cohorts and predose week 4 of the multiple-dose cohorts were combined in the week 4 timepoint assessment.
Efficacy evaluations: Mayo scores
Mayo scores34 were derived at week 4 and week 12 from disease diaries completed by patients throughout the study (stool frequency and rectal bleeding components), endoscopic examination by flexible sigmoidoscopy, and the physician's global assessment. Mayo scores were summarised as responder rates (proportion of patients with ≥3-point reduction and 30% improvement in total Mayo score, and ≥1-point decrease in rectal bleeding subscore or absolute rectal bleeding score of 0 or 1) and remission rates (proportion of patients with total Mayo score ≤2 points with no individual subscore exceeding 1 point). Endoscopic response was defined as ≥1-point improvement in endoscopic subscore, and endoscopic remission as endoscopic subscore of 0 or 1. The proportion of patients with complete mucosal healing (endoscopic subscore of 0) was also quantified.
Measurement of biomarkers
Feces and blood samples were collected predose and at intervals during 12–16 weeks' follow up, for measurement of faecal calprotectin, hsCRP, and α4β7-expressing lymphocytes. Fecal calprotectin was quantified by ELISA (ELISA, Calpro AS, Oslo, Norway), while plasma hsCRP was quantified using a standard in vitro diagnostic assay (Tina-Quant, Roche, Mannheim, Germany). Lymphocyte sub-populations expressing α4β7-integrin were determined in whole blood by 4-color flow cytometry using anti-α4 and anti-β7 antibodies (FACSCalibur and antibodies, Becton Dickinson, New Jersey, USA) and analysed with FloJo software (Tree Star Inc., Ashland, Oregon, USA).
Statistical methods
Sample sizes for this first-in-human study were selected to balance the need to minimise exposure of patients to PF-00547,659 with the need to provide adequate safety and tolerability information (primary outcome measure). The study was not powered to detect statistically significant differences in clinical/endoscopic response or remission rates, and biomarkers (secondary outcome measures).
Clinical response and remission rates were calculated for each PF-00547,659 dose strength (IV and SC routes combined) at weeks 4 and 12 to explore the dose–response relationship. Clinical response and remission rates were also calculated for the combined group of all PF-00547,659-treated patients, as were endoscopic response and remission rates. Only patients in the multiple-dose cohort were included in the week 12 analyses. Clinical and endoscopic response and remission rates were compared between PF-00547,659 (all dosages combined) and placebo groups, at weeks 4 and 12, using Fisher's exact test with a one-sided 5% significance level (SAS; Release 8.02, SAS Institute Inc., Cary, NC, USA). All patients who were randomised and received at least one dose of study treatment were included in efficacy analyses; patients who discontinued due to AEs or lack of efficacy were considered treatment failures and classed as non-responders.
Changes from baseline in natural log-transformed faecal calprotectin and plasma hsCRP levels were compared between PF-00547,659 and placebo at weeks 4 and 12, using an analysis of variance (ANOVA) model that allowed for variation in baseline levels. These results were back-transformed to give point estimates of the percentage changes and associated 95% CI. Change from baseline in CD4+ α4β7+ and CD4+ α4β7− lymphocytes was summarised using descriptive statistics (mean, SD) for patients receiving each PF-00547,659 dose strength and placebo, at weeks 4 and 12. For the CD4+ α4β7+ and CD4+ α4β7− lymphocyte analyses, PF-00547,659-treated patients were also subdivided into high-dose (1–10 mg/kg) and low-dose (0.03–0.3 mg/kg) groups.
Results
Patient demographics and disposition
Patients were enrolled at 17 sites in Europe from September 2005 to September 2008. In total, 80 patients were assigned to treatment following screening: 30 to single-dose cohorts; 50 to multiple-dose cohorts (figure 1). Twenty-one patients (70%) completed the single-dose study phase and nine (30%) discontinued: five due to lack of efficacy and four due to AEs not considered to be related to study drug. Thirty patients (60%) completed the multiple-dose phase of the study and 20 (40%) discontinued: 17 due to lack of efficacy and three due to AEs; one patient's AEs were considered to be related to the study drug (general physical health deterioration, frequent bowel movements and abnormal laboratory tests, on PF-00547,659 1 mg/kg SC). The majority of patients were white males (possibly due to entry criteria excluding women of childbearing potential at the start of the study; this was later amended due to low recruitment to allow also females of childbearing potential with adequate contraception), with similar distributions of demographic and baseline clinical characteristics across placebo and active treatment groups (table 1).
Demographic and baseline characteristics
Safety and tolerability
AEs were reported by similar proportions of patients on PF-00547,659 and placebo. The most common AEs affected the gastrointestinal system, including abdominal pain or tenderness; few cases were considered treatment related (table 2). Most AEs were mild or moderate in intensity.
Adverse events occurring in ≥2 patients in placebo or PF-00547,659 combined groups (single- and multiple-dose study phases combined)
There was no evidence of opportunistic infections arising during PF-00547,659 treatment, within or outside the gastrointestinal system. Overall incidence of gastrointestinal tract infections was low; five patients receiving PF-00547,659 and one receiving placebo reported enteritis/gastroenteritis, but none of the cases were considered by investigators to be related to study treatment. Respiratory tract infections were infrequent. One case (moderate upper respiratory tract infection) was considered potentially related to PF-00547,659 treatment, although the patient was also taking azathioprine, which could have increased susceptibility. Few AEs affecting the central nervous system (CNS) (mostly dizziness) were reported, with the majority of cases not considered to be treatment related.
There were no deaths during the study. Three patients reported nine serious AEs (SAEs) within the 16-week study (one patient in each of the placebo, 3 mg/kg IV and 1 mg/kg SC multiple-dose groups). Only the patient receiving PF-00547,659 1 mg/kg SC had SAEs that were investigator-assessed as potentially related to treatment: rectal haemorrhage, general physical health deterioration, joint (ankle) abscess, wound infection (abdominal wall wound from proctocolectomy undertaken upon worsening of ulcerative colitis), and abnormal laboratory tests (elevated leucocyte count; low haemoglobin); this patient discontinued the study. SAEs were recorded for six more patients at follow-up visits after study end; these AEs were considered to be related to underlying ulcerative colitis or other illnesses. Laboratory test abnormalities did not suggest any treatment- or dose-related patterns in changes in any parameter.
There was no evidence of an immunogenic response to the drug as no anti-drug antibodies were detected in the presence of the drug, or 1 month after the last injection, and no injection site reactions were observed. Measurements at later time points after complete disappearance of the drug would be needed to definitively rule out anti-drug antibodies, but these samples are not available.
Efficacy results
Mayo scores
Overall response rates were higher with PF-00547,659 than placebo at both week 4 (52% vs 32%, respectively; p=0.102) and week 12 (42% vs 21%, respectively; p=0.156; multiple-dose cohort only), but these differences did not reach statistical significance (figure 2A). Remission rates were similar in PF-00547,659 and placebo groups at week 4 (13% and 11%, respectively), but it may take longer to achieve full remission than simply response, and by week 12 remission rates had risen to 22% with PF-00547,659, while no patients on placebo remained in remission (figure 2B). The difference in week 12 remission rates between PF-00547,659 and placebo approached, but did not reach statistical significance (p=0.056). When separate PF-00547,659 dose groups were considered, there was no clear pattern of increasing response or remission rate with increasing PF-00547,659 dose (figure 3). Endoscopic response and remission rates were numerically, but not statistically higher, at both timepoints, with PF-00547,659 than placebo (figure 2C,D). Although complete mucosal healing rates were similar with PF-00547,659 and placebo at week 4 (10% (6/60) vs 11% (2/19), respectively), by week 12, 14% (5/36) of patients on active treatment had complete mucosal healing compared with 0% (0/14) of those on placebo. Examination of individual Mayo component scores indicated that a reduction in rectal bleeding, as well as endoscopic improvement/healing, contributed towards the improvements in overall Mayo scores seen with PF-00547,659.

Improvement in signs and symptoms of ulcerative colitis with PF-00547,659: overall clinical response (*) and remission (†) rates and endoscopic response (‡) and remission (§) rates based on Mayo scores at week 4 and 12. (A) ≥3-point reduction and 30% improvement in total Mayo score (all components including endoscopy ≥1 point), and a decrease in rectal bleeding subscore, or absolute rectal bleeding score of 0 or 1. (B) Total Mayo score ≤2 points with no individual subscore >1 point. (C) ≥1-point improvement in Mayo endoscopic subscore. (D) Mayo endoscopic subscore of 0 or 1. p Values obtained using one-sided Fisher's exact test to test the probability that active treatment is associated with higher response/remission rates than placebo. Week 4 data include all patients in single-dose cohort and data for assessments following the first dose for all patients in multiple-dose cohorts. Week 12 data include patients in the multiple-dose cohort only.

Clinical response (A) and remission (B) rates (based on total Mayo score) at weeks 4 and 12 by PF-00547,659 dose.
Biomarkers
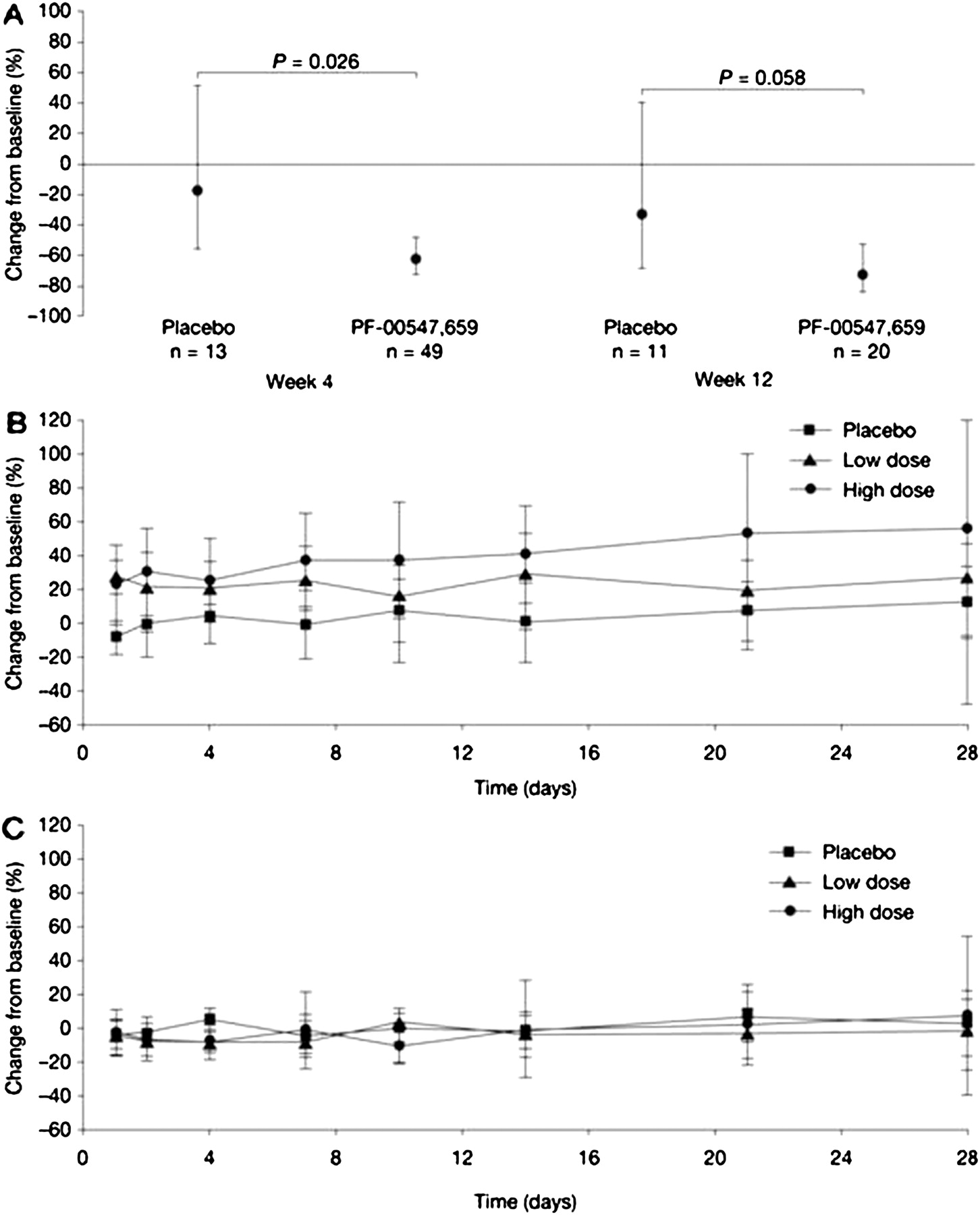
The reduction in mean faecal calprotectin from baseline was significantly higher with PF-00547,659 than with placebo at week 4 (63% vs 18%, respectively) and of borderline significance at week 12 (73% vs 34%, respectively) (figure 4A). At the individual patient level, there was a trend for a high proportion of patients in the PF-00547,659 group to have reductions in faecal calprotectin from baseline to week 4 or week 12 (data not shown). In the placebo group, individual patient responses were highly variable and there was no clear trend for a reduction in faecal calprotectin from baseline at either timepoint.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in biomarkers following dosing with PF-00547,659 or placebo: faecal calprotectin (A); CD4+ α4β7-positive lymphocytes (B); CD4+ α4β7-negative lymphocytes (C). (A) Baseline adjusted geometric mean and associated 95% CI are presented as a percentage change from baseline. (B and C) Descriptive statistics only. N numbers were variable over the 28 days.
Clinically meaningful changes in plasma hsCRP could not be detected as baseline levels were close to normal values (table 1). Individual changes in plasma hsCRP from baseline with both PF-00547,659 and placebo were also highly variable, with no significant differences in mean values between the two treatments at week 4 (−14% vs −28%, respectively) or 12 (−57% vs 7%, respectively).
There was a trend for an increase from baseline in the number of circulating CD4+ α4β7+ lymphocytes in patients receiving PF-00547,659, but not in patients receiving placebo (table 3); however, this was not statistically significant due to a large inter-subject variability. The effect was sustained at 4 weeks in both the high-dose (1–10 mg/kg) and low-dose (0.03–0.3 mg/kg) PF-00547,659 groups (figure 4B). The mean number of CD4+ α4β7− cells in the high- and low-dose PF-00547,659 groups, and the placebo group, remained close to baseline values throughout the study (figure 4C). For both cell types, individual responses were subject to considerable variation.
Mean change in CD4+ α4β7-positive lymphocytes from baseline in whole blood following dosing with PF-00547,659 or placebo
Discussion
Blocking MAdCAM is a rational mechanism of action for therapeutic intervention in IBD. MAdCAM represents an organ-specific target, as expression is restricted mainly to gastrointestinal tract mucosa.22 This organ specificity may translate to a favourable safety profile; in particular, the absence of MAdCAM in the brain microvasculature32 suggests that drugs acting on MAdCAM would not impact the body's ability to respond to CNS infections such as PML.
Indeed, this first-in-human trial of PF-00547,659 had a relatively low incidence of non-gastrointestinal AEs. Gastrointestinal AEs were common in both PF-00547,659 and placebo groups, owing to the disease under study, and most AEs were not considered to be treatment related. The majority of SAEs occurred after follow-up and were related to underlying disease rather than study drug. While nine patients (30%) discontinued the single-dose phase and 20 (40%) discontinued the multiple-dose phase of the study, only seven withdrew due to AEs. Moreover, only one of these patient's AEs were considered to be related to the study drug. Overall, PF-00547,659 had an AE profile similar to placebo, and no safety concerns emerged in laboratory tests or other safety monitoring.
Biological therapy is often associated with immunogenic reactions, development of antidrug antibodies, which diminish treatment response and can lead to infusion/injection site reactions. PF-00547,659 is a fully human monoclonal antibody developed using Abgenix XenoMouse™ technology (Amgen, Thousand Oaks, California, USA), a platform that has yielded antibodies with low intrinsic immunogenicity. In this study, no antidrug antibodies were detected during treatment up to week 12 for all patients, and no injection site reactions were observed. Longer-term studies, involving a much larger number of patients, are required to confirm a lack of immunogenicity with prolonged treatment (administered at clinically relevant doses) and assess other aspects of long-term safety, including risk of opportunistic infections, which only tend to become evident with extended treatment. Biological agents for IBD are likely to be subject to scrutiny in this regard following cases of PML with long-term natalizumab therapy. Up to 80% of adults carry the JC virus in a latent form, after being exposed during childhood. The virus resides in the kidneys, bone marrow and the gastrointestinal tract and re-activation, most likely via the haematogenic route to the brain, causes PML. It is not clear if defective homing of cytotoxic T cells to the brain or the appearance of more virulent JC virus in the brain contribute to the risk of PML as seen after natalizumab therapy.
This study investigated the preliminary efficacy of PF-00547,659 in patients whose ulcerative colitis is not controlled with conventional therapy and found that, within small sample size limitations, PF-00547,659 produced some potential benefits over placebo on clinical and endoscopic endpoints. However, no statistical differences between PF-00547,659 and placebo were reported for any efficacy endpoints. Overall Mayo responder rates were approximately 40–50% with PF-00547,659. No clear dose–response relationship was observed with PF-00547,659, although small groups (most doses administered to four patients) and limited sensitivity of the clinical and endoscopic endpoints confounded full exploration of dose–response relationships within this initial study. The high discontinuation rate due to lack of efficacy (22/80) was not surprising for a patient population with a fairly extensive, multidrug treatment history and advanced disease that was not adequately controlled with conventional medications.
Results of biomarker assays supported the clinical findings of this study. Levels of faecal calprotectin, which reflects disease activity and is a good predictor of disease course in ulcerative colitis,35–37 declined in patients treated with PF-00547,659, and were significantly reduced with PF-00547,659 relative to placebo at week 4. An increase in the number of CD4+ α4β7+ lymphocytes following PF-00547,659 administration, with no change in CD4+ α4β7− cell numbers, was consistent with MAdCAM blockade, and reflects the expected pharmacology that has been reported in primate studies with PF-00547,659.33 It should be noted, nonetheless, that individual lymphocyte responses were subject to huge variation. Lack of change in α4β7− cell numbers provided validation for the sensitivity of the assay to any potential drug-related effects. As mean hsCRP levels were close to normal levels at baseline and post-treatment changes were subject to considerable variation, hsCRP was not considered a useful biomarker for ulcerative colitis inflammation in this study.
This first-in-human study was conducted in patients with ulcerative colitis, but the drug's mechanism, blocking MAdCAM to attenuate lymphocyte migration to the gut mucosa, would also be expected to be effective in Crohn's disease. The next steps in development of the compound for either indication should include fully powered dose-ranging studies, this time applying an even gender distribution (which was skewed in the present study due to the initial exclusion of women of child-bearing potential). Dose selection for such studies can be guided by pharmacokinetic profiles, which indicate that MAdCAM becomes fully saturated at PF-00547,659 doses of 1 mg/kg or higher,38 and levels of CD4+ α4β7+ lymphocytes, which showed some differentiation of dose effect. Longer-term studies are needed, to assess efficacy in maintenance as well as induction of remission, and to confirm the apparently positive safety profile observed in this study.
In conclusion, the favourable short-term (typically 12–16-week) safety profile and preliminary efficacy results observed with the MAdCAM antibody PF-00547,659 in this first-in-human study pave the way for a further longer-term investigation of clinically relevant doses in larger numbers of patients, as will be necessary to establish the potential role of PF-00547,659 in the treatment of IBD.
References
Footnotes
Writing assistance: Editorial support with the drafting of this manuscript was provided by Samantha Stanbury PhD and Louise Norbury MSc (FireKite, UK), and funded by Pfizer Inc.
Data analysis was led by Jacqueline Spanton, Steven W. Martin and Wojciech Niezychowski at Pfizer Ltd.
Funding This study was sponsored by Pfizer Inc.
Competing interests SV has received consulting fees (<$10,000/year) from Schering-Plough, speaker fees (<$10,000/year) from Schering-Plough, UCB and Ferring, and grant support (€65,000) from UCB. JFD has received speaker fees (<$10,000/year) from Ferring and Roche. AL has received consulting fees (<$10,000/year) from UCB, speaker fees (<$10,000/year) from Abbott, Essex and the Falk Foundation, and grant support from Wolff Pharma ($80,000 in 2008), IZKF (institutional research grant of $200,000 covering the period 2006–2008) and the German Research Foundation (DFG; $70,000 over 2008/9). JP has received consulting fees (<$10,000/year) from Schering-Plough, Abbott and Ferring, speaker fees (<$10,000/year) from Schering-Plough and Abbott, and grant support from the Spanish Ministry of Science and Innovation ($175,000 over 2007/9) and Schering-Plough ($125,000 over 2008/9). SG has received speaker fees (<$10,000/year) from Schering-Plough, Centocor, UCB and Abbott, speaker fees (<$10,000/year) from Schering-Plough, Procter and Gamble and (>$10,000/year) Abbott, and grant support ($90,000 over 2006–2007) from Schering-Plough. WN has equity/stock ownership (>$10,000) in Pfizer Ltd and Johnson & Johnson, and is an employee of Pfizer Ltd. GB and J Spanton have equity/stock ownership (>$10,000) in Pfizer Ltd and are employees of Pfizer Ltd. SWM has equity/stock ownership (>$10,000) in Pfizer Ltd and Amgen, and is an employee of Pfizer Ltd. US has no financial disclosures to declare. J Sirotiakova has no competing interests.
Patient consent Obtained.
Ethics approval The research protocol was approved by the appropriate institutional review board or independent ethics committee for each study site. All participants gave written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.