


Abstract
Sister of P-glycoprotein (SPGP), a novel murine cDNA and member of the ATP-binding cassette superfamily highly homologous to P-glycoprotein (Pgp), was cloned. Moreover, its genomic clone was isolated and localized to chromosome 2 by fluorescence in situ hybridization. SPGP was functionally evaluated relative to MDR1 after subcloning SPGP cDNA into a retroviral bicistronic vector capable of expressing both SPGP and the green fluorescent protein. LLC-PK1 and MDCKII cells were transduced with this retrovirus and SPGP-positive clones were isolated. Drug uptake and efflux was compared in cells ectopically expressing either SPGP or human MDR1. SPGP cells had decreased uptake of taurocholate and vinblastine compared with LLC-PK1 cells. Additional studies revealed that vinblastine efflux was accelerated by SPGP compared with LLC-PK1. Further comparison revealed that although MDR1 easily impaired uptake of vincristine, daunomycin, paclitaxel, and digoxin, SPGP had no effect on uptake of these drugs. However, further studies demonstrated that, like MDR1, SPGP effluxed calcein-acetoxymethyl ester (AM). Unlike MDR1, SPGP was incapable of effluxing rhodamine 123. Although cyclosporine A and reserpine blocked calcein-AM transport by MDR1, these drugs had either minimal or no effect, respectively, on blocking SPGP efflux of calcein-AM. In contrast, ditekiren, a linear hexapeptide, readily and preferentially inhibited SPGP efflux of calcein-AM. Further studies with three structural analogs of ditekiren revealed that one analog inhibited SPGP efflux of calcein-AM, although not as potently as ditekiren. These are the first studies to reveal that SPGP has distinct transport properties compared with MDR1.
A novel member of the P-glycoprotein (Pgp) family called sister P-glycoprotein (SPGP) was first identified in pig liver from a partial cDNA sequence (Childs et al., 1995). SPGP belongs to the ATP-binding cassette (ABC) transporter family and is closely related to the multidrug resistance gene (MDR1) and its protein product Pgp. Pgp transports a wide variety of structurally dissimilar drugs, and overexpression of MDR1/Pgp contributes to multidrug resistance to cancer chemotherapeutic agents. Because Northern blot analysis demonstrated the gene product is expressed exclusively in liver (Childs et al., 1995), it was suggested that the SPGP transporter might have a role in efflux of endogenous compounds (bile acids) and exogenous compounds (drugs) into the bile. A recent study demonstrated that the rat SPGP expressed in insect cells had the capability of transporting bile acids (Gerloff et al., 1998). However, it is currently unknown whether the rodent SPGPs possess drug transport capability. Moreover, we do not know if murine SPGP and MDR1/Pgp will overlap with regard to drug substrates. SPGPs sequence similarity to Pgp and the finding that mice nullizygous for the drug-transporting Pgps (mdr1a andmdr1b) still transport digoxin (Mayer et al., 1997; Schinkel et al., 1997a) and paclitaxel (Schinkel et al., 1997a) from the liver suggests that another protein(s) located at the bile canalicular membrane is involved in transporting these drugs.
Because Pgp and SPGP are both expressed in the liver, it is clear that the extent of overlap between Pgp and SPGP drug substrates and inhibitors needs to be established to judiciously predict the role SPGP plays in drug disposition in vivo. In addition, because SPGP is preferentially expressed in the liver, the extent to which cholestatic drugs interact with SPGP needs to be defined. There have been reports of drugs and steroids causing cholestasis and microtubule-disrupting agents have been reported to produce a bile salt-dependent hepatotoxicity (Katagiri et al., 1992; Crocenzi et al., 1997)—potentially by interacting with SPGP-mediated transport. Further studies have revealed that some vinca alkaloids have the potent ability to interfere with canalicular transport processes; one could speculate that this effect may be partly attributed to a functional interaction with SPGP (Watanabe et al., 1992). To assess the drug transport function of murine SPGPs, we stably introduced the murine SPGP into LLC-PK1 cells using a novel retrovirus. Transport function was evaluated in SPGP cells compared with either LLC-PK1 cells or LLCPK MDR1 cells stably expressing the human MDR1 cDNA because MDR1 is the closest homolog to murine SPGP. We demonstrate for the first time that SPGP, with respect to both substrates and inhibitors, is a unique and functionally distinct transporter compared with MDR1.
Experimental Procedures
Materials.
[3H]taurocholic acid (2 Ci/mmol) and [3H]paclitaxel (2.4 Ci/mmol), and [3H]vinblastine-H2SO4(7.6 Ci/mmol) were obtained from NEN Life Science Products (Boston, MA) and Moravek Biochemicals (Brea, CA), respectively. Taurocholic acid, vinblastine, and paclitaxel were purchased from Sigma (St Louis, MO). The 1.1-kilobase (kb) murine SPGP expressed sequence tag (EST) (aa066341.gb_est1 from genbank) was purchased from Genome Systems (St. Louis, MO) and confirmed by DNA sequence analysis (see below). This EST clone is hereafter referred to as the 1.1-kb fragment of the mouse SPGP cDNA. Ditekiren and its analogs were obtained from the Upjohn Co. (Kalamazoo, MI). Calcein-acetoxymethyl ester (AM) was purchased from Molecular Probes (Eugene, OR). Rhodamine 123 was purchased from Sigma (St. Louis, MO).
Cell Lines.
The human embryonic kidney 293T cells (generous gift of the laboratory of Dr. A. Nienhuis, St. Jude Children's Research Hospital, Memphis, TN) and the Madin-Darby canine kidney cells strain II (MDCKII), a polarizing epithelial cell line derived from proximal tubule, were maintained in Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal calf serum (heat inactivated serum for the 293T cells), 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2 humidified atmosphere. The MDCKII and LLC-PK1 cells overexpressing the SPGP (MDCKII Sis2, MDCKII Sis5, LLCPKSis2, and LLCPKSis7) were used between passage numbers 1 and 15. The MDCKII MDR1 cell lines and the LLC-PK1 derivative cell lines containing human MDR1 (LLCPK MDR1) were kindly provided by Drs. Raymond Evers and Alfred Schinkel, respectively (The Netherlands Cancer Institute, Amsterdam, The Netherlands) and have been described previously (Bakos et al., 1998).
Screening and Isolation of the cDNA Clones.
An adult mouse BALBc liver cDNA library was obtained from Clonetech (Palo Alto, CA). Approximately 800,000 phages from this cDNA library were hybridized with a [32P]dCTP-labeled mouse SPGP EST. Hybridization was carried out for 20 h at 42°C under standard hybridization conditions [50 mM sodium phosphate, pH 6.5, containing 50% formamide, 5× standard saline citrate (0.15 M NaCl and 0.015 M trisodium citrate), 5× Denhardt's solution, 0.1% SDS, and 100 μg/ml sheared salmon sperm DNA]. After high-stringency washing (0.1 SSC/0.1% SDS) at 52°C, 168 positives clones were picked. Sequence information was used to design SPGP-specific oligonucleotide primers to screen these clones by polymerase chain reaction (PCR). The largest cDNA insert (4.7 kb) identified was plaque-purified as described previously (Schuetz et al., 1989). Sequencing reactions were performed on double-stranded plasmids by the Center for Biotechnology at St Jude Children's Research Hospital using dye-terminator cycle sequencing “ready reaction” kits containing AmpliTaq DNA polymerase FS (Biosystems, Inc., Foster City, CA) and synthetic oligonucleotides. Reactions were analyzed on PE/ABI model 373 sequencers.
In Vitro Translation.
The 4.7-kb fragment was subcloned into the mammalian expression vector pcDNA3 (InVitrogen, Carlsbad, CA) and orientation was confirmed by sequence analysis. The in vitro translation was performed with the TNT Coupled Reticulocyte Lysate Systems (Promega, Madison, WI) according to the manufacturer's instructions. The reaction products were fractionated on a 7.5% denaturing polyacrylamide gel. The gel was fixed and the products were visualized by autoradiography.
Northern Blot Analysis.
A mouse multiple-tissue Northern blot (Clontech, Palo Alto, CA) [2 μg of poly(A+) RNA per lane] was hybridized with the 1.1-kb fragment of the mouse SPGP cDNA and a human β-actin probe labeled with a [32P]dCTP by random priming (Stratagene, La Jolla, CA) at 42°C overnight. Total RNA from the cell lines was extracted and 10 μg were analyzed by Northern blotting as described previously (Schuetz et al., 1995b). Before transfer, the RNA integrity was assessed by ethidium bromide staining. After transfer, the nylon membrane was hybridized with the 1.1-kb cDNA fragment of the mouse SPGP labeled as described previously.
Mouse SPGP Gene Analysis.
A murine bacterial artificial chromosome (BAC) library was screened by hybridization with the 1.1-kb fragment of the mouse SPGP cDNA (Genome Systems, Inc.) and four positive clones were identified. We identified SPGP exons in one BAC (clone 1) by subcloning, hybridization, and sequence analysis. Exon sequence primers were designed to determine intron lengths. PCR reactions were performed with the Expand High Fidelity PCR System (Boehringer Mannheim, Indianapolis, IN). PCR products were analyzed on a 0.8% agarose gel.
Chromosomal Localization.
Fluorescence in situ hybridization (FISH) was performed by labeling the BAC (clone 1) DNA with digoxigenin dUTP. A probe specific for the telomeric region of chromosome 2 was also labeled. Labeled probes were combined with sheared mouse DNA and hybridized to metaphase chromosomes derived from mouse embryo fibroblast cells (that are hyperdiploid) in a solution containing 50% formamide, 10% dextran sulfate, and 2× SSC. Specific hybridization signals were detected by incubating the hybridized slides with fluorescein-conjugated antidigoxigenin antibody followed by counterstaining with 4,6-diamidino-2-phenylindole.
Development of SPGP Antibody.
We selected a 15-amino acid peptide (KGAYYKLVITGAPIS) from the carboxyl terminus (position 1307–1321) of the mouse SPGP cDNA. Polyclonal antisera were produced by immunization of two rabbits with the SPGP peptide coupled to keyhole limpet hemocyanin and complete Freund's adjuvant (Rockland Laboratories, Inc., Gilbertsville, PA). The SPGP antiserum was purified by peptide affinity chromatography (Research Genetics, Huntsville, AL).
Preparation of GST-SPGP Fusion.
The cDNA fragment of mouse SPGP representing amino acids 1157 to 1321 (predicted molecular mass, 18.08 kDa) was subcloned into pGEX5x1 (Pharmacia, Piscataway, NJ) and Escherichia coli BL21 cells were transformed with this recombinant. The expected size of this glutathione transferase (GST)-SPGP fusion is approximately 44 kDa. The bacterial cells were grown overnight in 2xYTG containing 2% glucose. The cultures were then diluted and grown at 32°C. A 2-h incubation in isopropyl β-d-thiogalactopyranoside (0.1 mM) produced maximal recombinant SPGP induction. The fusion protein was run on a 10% denaturing polyacrylamide gel and transferred to nitrocellulose filters. Filters were blocked in a 1× PBS, 0.1% Tween-20, and 10% milk buffer and immunoreacted first with a goat anti-GST-fusion antibody followed by peroxidase-conjugated antigoat IgG, and finally with peroxidase-conjugated antirabbit IgG. The blots were developed with the Amersham enhanced chemiluminescence detection system (Amersham, Arlington Heights, IL). Subsequently, the same blot was stripped by a 30-min incubation in 0.2 M glycine, pH 2.8, and reprobed with antiserum to SPGP (see below).
Immunoblot Analysis.
Crude membranes were prepared from mouse liver as described previously (Schuetz et al., 1995a). Protein was estimated by the Bio-Rad (Hercules, CA) protein assay using bovine serum albumin as standard. The crude membrane proteins (300 μg) were resuspended in standard Laemmli sample preparation buffer and loaded onto a 7.5% denaturing polyacrylamide gel and transferred to nitrocellulose filters. The filters were blocked in a 1× PBS containing 0.1% Tween-20 and 10% nonfat dry milk, immunoreacted with polyclonal rabbit anti-SPGP IgG followed by peroxidase-conjugated antirabbit IgG, and then developed with the Amersham enhanced chemiluminescence detection system.
Generation of SPGP Stable Cell Lines.
The mouse SPGP cDNA was cloned into the MSCV-IRES-GFP vector (kindly provided by Dr. Robert Hawley, Holland Laboratory, American Red Cross, Rockville, MD) using the EcoRI site. 293T cells were cotransfected with 10 μg each of MSCV-SPGP-IRES-GFP and the helper plasmid pEQEco by standard calcium phosphate precipitation (Persons et al., 1998). Forty-eight hours after the transfection, the supernatant was collected, filtered, titered, and frozen at −80°C. To confirm transfection, the 293T cells were analyzed for green fluorescence protein (GFP) expression. LLC-PK1 cells were transduced with MSCV-SPGP-IRES-GFP using the helper plasmid pSRα-G (Yang et al., 1995) and pEQPAM3-e (kindly supplied by P. Kelly and E.F. Vanin, Department of Hematology/Oncology, St Jude Children's Research Hospital, Memphis, TN). MDCKII cells were transduced with MSCV-SPGP-IRES-GFP using the helper plasmid pEQPAM3-e (Persons et al., 1998). Briefly, the cells were plated at 5 × 104 cells/60 mm tissue culture dishes and then medium replaced by the retroviral supernatant supplemented with 6 μg/ml polybrene and placed overnight in the incubator at 37°C in a 5% CO2 humidified atmosphere. The transduction was performed a total of four times. The transduced cells were expanded and the GFP-positive cells were selected after fluorescence-activated cell-sorting assay. Subsequently, limiting dilution was used to select individual cell clones expressing varying amounts of SPGP. For each clone, a total lysate was prepared and loaded on a denaturing polyacrylamide gel for SPGP detection by Western blot. For both cell types, the retention of SPGP in individual clones was monitored periodically by assessing GFP or SPGP by either immunofluorescence or western blot analysis. We found that SPGP expression was stable for at least 15 passages.
Drug Accumulation.
To assess drug uptake, we used a modification of the procedure previously described (Schuetz and Schuetz, 1993). Briefly, cultured cells were placed at 37°C in media containing 3H substrates at the indicated concentrations: taurocholate (1 mM) or vinblastine (0.5 μM), daunomycin (2 nM), vincristine (1.0 μM), paclitaxel (1.0 μM), digoxin (1.0 μM), tamoxifen (1.0 μM). At the time intervals shown in the figure, individual dishes were washed three times with ice-cold PBS, cells scraped to harvest, resuspended in PBS, and sonicated and analyzed for radioactivity using a scintillation counter. To determine the kinetic properties of vinblastine, cultured cells were incubated with varying vinblastine concentrations and processed as described above. The radioactivity associated with the cells is expressed as either picomoles per 106 cells or picomoles per microgram of protein. Each point is the mean ± SD of at least three independent experiments with duplicate determinations (Fig. 7). K m andV max values were calculated using a modified form of the Michaelis-Menten equation.K m is the concentration that gives half the maximal accumulation of vinblastine in the cell;V max is the maximal concentration of vinblastine accumulation in cells.

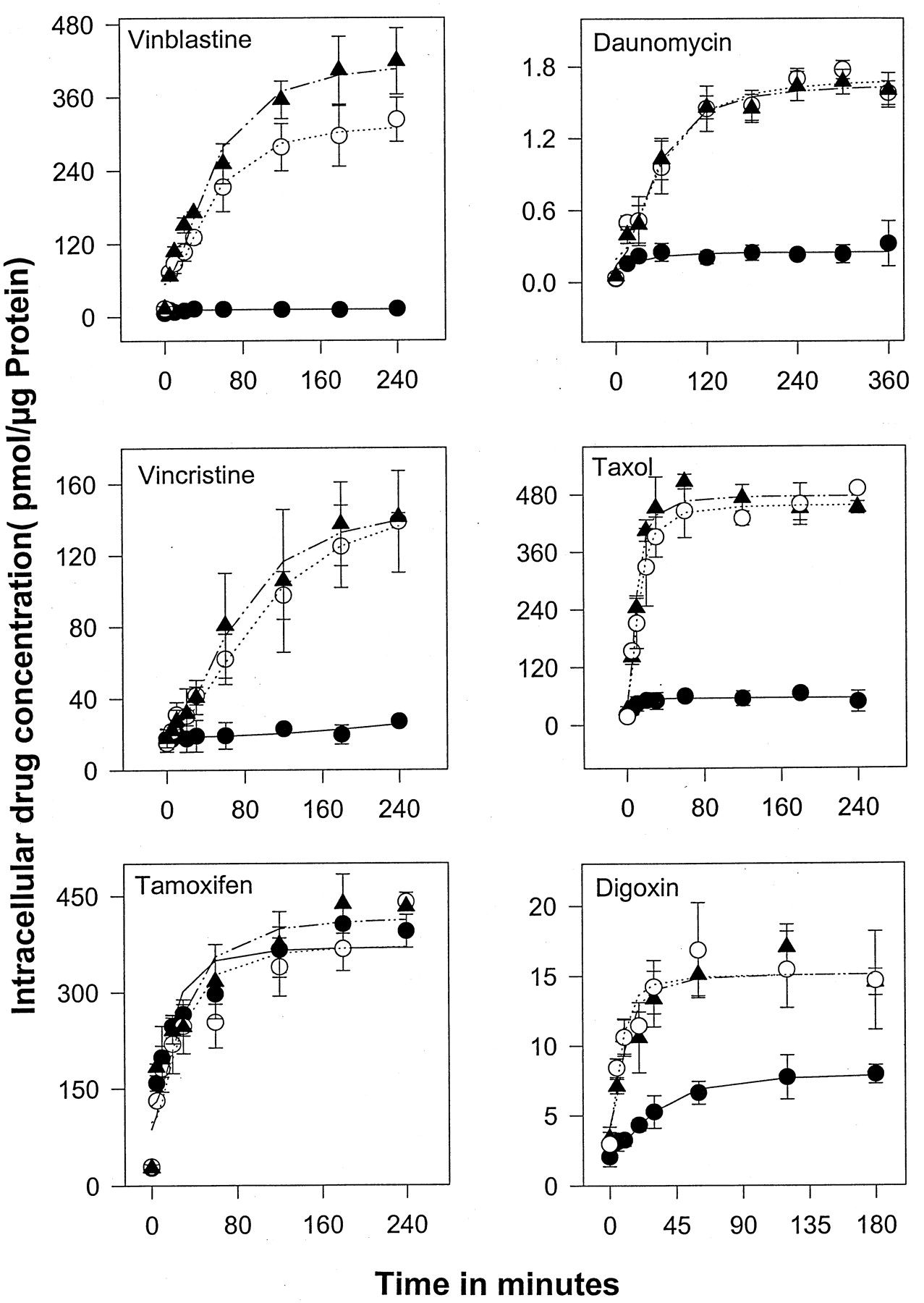
Uptake of vinblastine, vincristine, tamoxifen, daunomycin, paclitaxel, and digoxin in SPGP compared with MDR1/Pgp cells. Cells were incubated in medium containing3H-radiolabeled drugs for varying intervals at 37°C. The3H-radiolabeled drugs associated with the cells were determined by scintillation counting and expressed as picomoles pere microgram of protein. Each point is the mean ± S.D. of at least three independent experiments performed in duplicate. ●, LLCPK MDR1; ○, LLCPKSis7; ▴, LLC-PK1
Drug Efflux.
Drug efflux was performed essentially as described previously (Schuetz and Schuetz, 1993). Cells were incubated for 2 h at 37°C with 1 μM [3H]paclitaxel or 1 μM [3H]vinblastine. Cells overexpressing Pgp (MDCKII MDR1 and LLCPK MDR1) were incubated with 5 μM [3H]vinblastine. Drug accumulation was terminated by washing three times with ice-cold PBS. This procedure removed over 99% of the extracellular radioactivity. At this point, some culture dishes were used to determine total cellular bound radioactivity by scraping the cells, sonicating, and then determining radioactivity by liquid scintillation counting. Parallel cultures were incubated again in 1 ml of drug-free medium at 37°C. Efflux of drug was assessed by removal of a 50-μl aliquot of medium every 20 min during the first hour and every hour for 4 h. Each aliquot was analyzed for radioactivity. The proportion of cellular radioactivity released into the medium was then determined by multiplication by a factor of 20 and expressing this value as a proportion of the initial intracellular bound radioactivity. Each point is the mean ± S.E.M. of at least three independent experiments (Figs. 5 and 6).

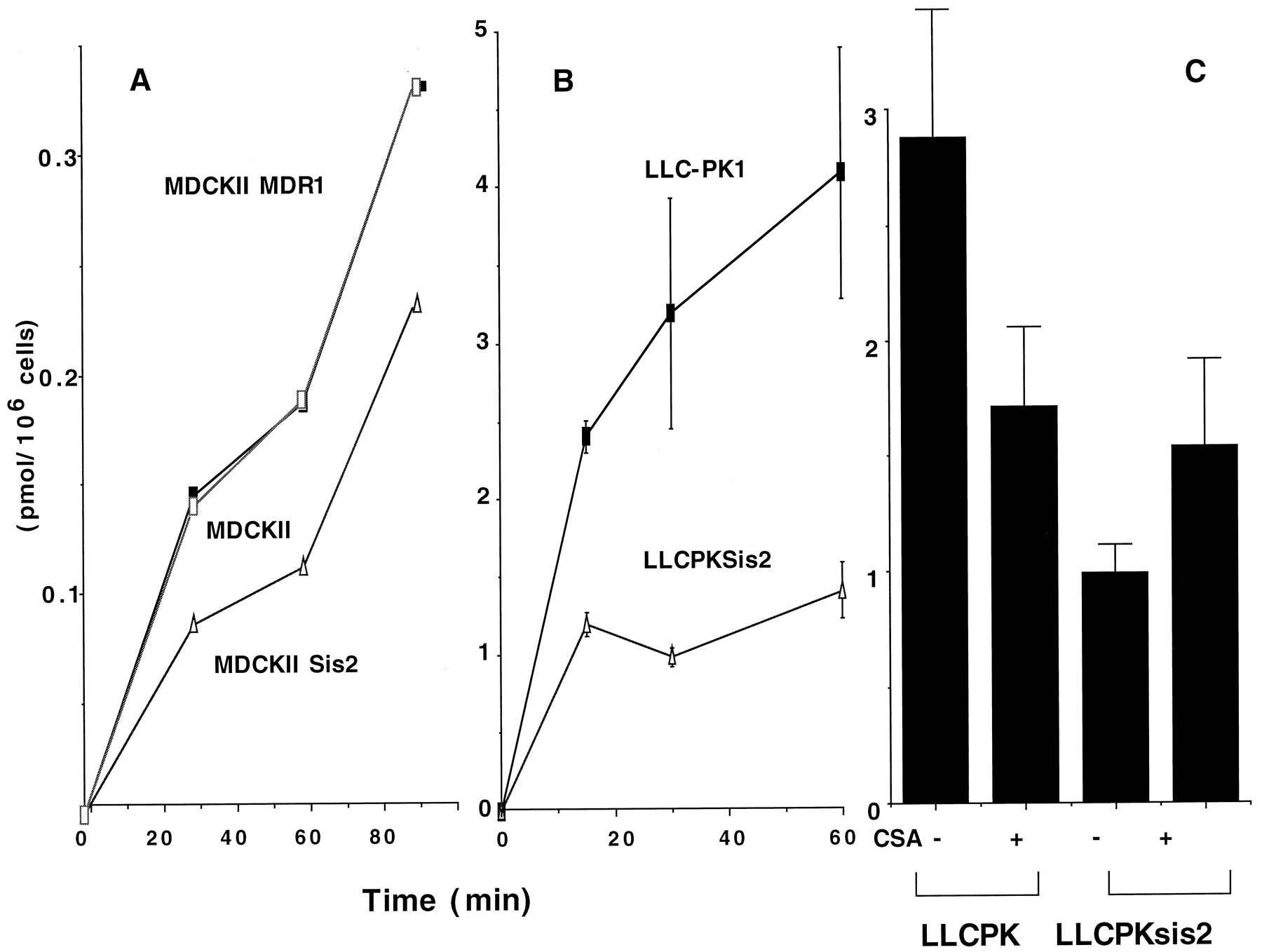
Taurocholate uptake in MDCKII, LLC-PK1, MDRI, and SPGP cells. MDCKII, MDCKII, MDR1, and MDCKII Sis2 (A) and LLC-PK1 and LLCPKSis2 (B) cells were incubated in media containing 1 mM [3H]taurocholate for 15, 30, 60, or 90 min at 37°C, and [3H]taurocholate associated with the cells was determined by scintillation counting and expressed as picomoles per 106 cells. Each point is the mean ± S.E.M. of at least three independent experiments. C, LLC-PK1 or LLCPKsis2 cells were incubated in media containing 1 mM [3H]taurocholate for 60 min at 37°C in either the presence or absence of 10 uM CsA and [3H]taurocholate associated with the cells was determined by scintillation counting and expressed as picomoles per 106 cells. Each point is the mean ± S.E. of at least three independent experiments.

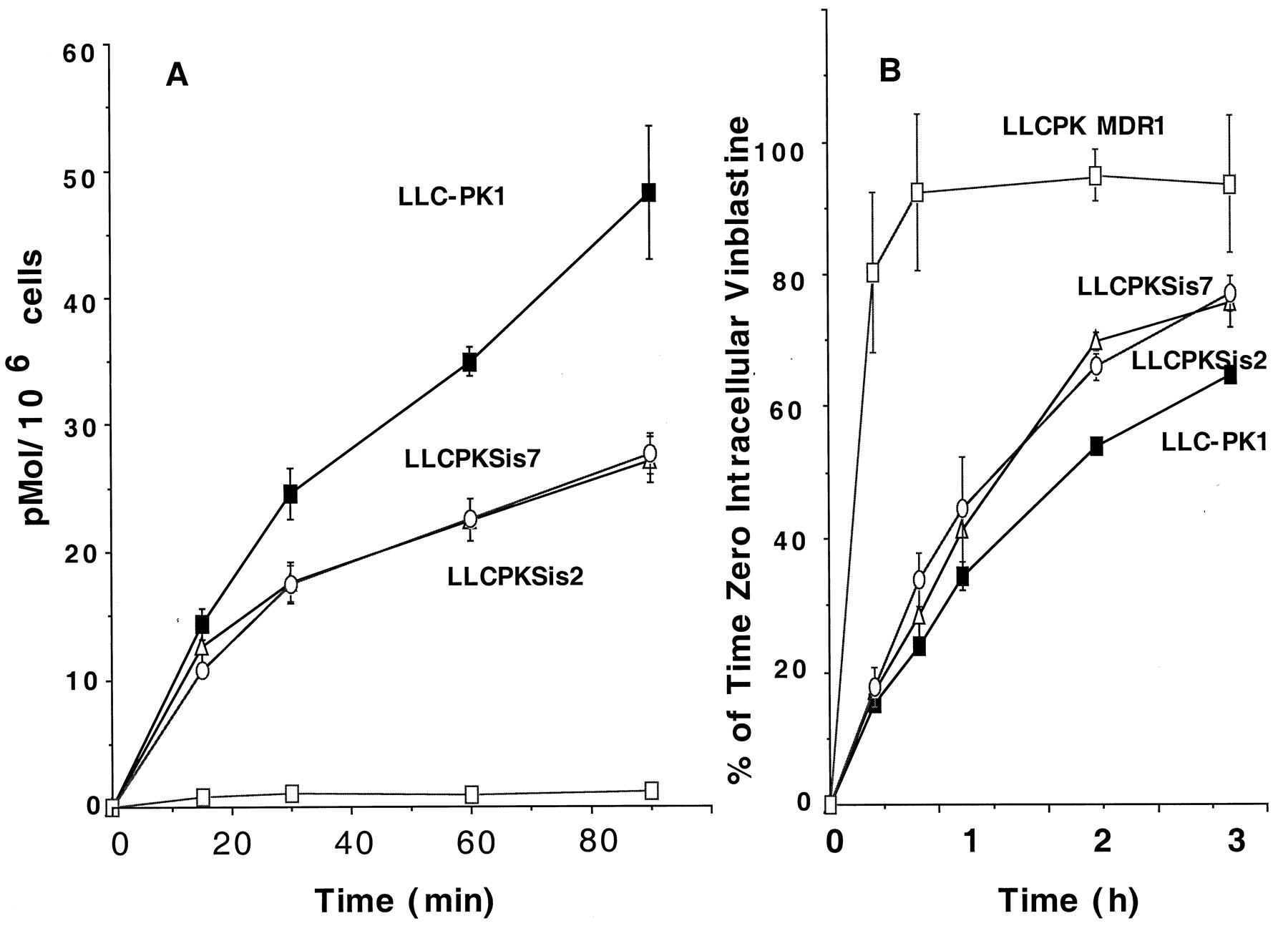
Vinblastine uptake and efflux in LLC-PK1, LLCPKSis2, LLCPKSis7, and LLCPK MDR1 cells. A, drug uptake: cells were incubated in medium containing 0.5 μM [3H]vinblastine for 15, 30, or 60 min at 37°C, and [3H]vinblastine associated with the cells was determined by scintillation counting and expressed as picomoles per 106 cells. B, drug efflux: cells were incubated in medium containing 1 μM [3H]vinblastine for 2 h (MDR1 cells were incubated with 5 μM drug), washed three times with ice-cold PBS, and some cells were assayed for cell-associated radioactivity (0 h). Other cells were incubated again in a drug-free medium at 37°C and, at the indicated times, an aliquot of 50 μl was removed and the proportion of radioactivity in the medium expressed as a percentage of the initial amount (0 h) of intracellular drug. Each point is the mean ± S.E. of at least three independent experiments.
Calcein-AM Fluorometry Assay.
Cells were cultured in Costar 96-well plates (Fisher, Pittsburg, PA) on day zero at 100,000 cells/well in medium. On day 1, medium was removed and the wells were washed once with 200 μl of PBS. One hundred microliters of PBS was added in each well, and just before the experiment, 100 μl of PBS containing calcein-AM (2 μM) was added to reach the final calcein-AM concentration of 1 μM. The microplates were analyzed at room temperature with a fluorescence microplate reader (Cytofluor 2350; Millipore, Bedford, MA) with excitation and emission wavelengths set at 485 nm and 530 nm, respectively. The plate was scanned for 3-min intervals repeated 11 times over 30 min at room temperature. Comparison of individual experiments was performed after normalization. Basal calcein fluorescence (time zero) was determined for each cell line after subtraction of the fluorescence from cells incubated in the absence of calcein-AM. Next, intracellular calcein fluorescence was determined at each time point and divided by the basal value and expressed as the fold increase in intracellular calcein fluorescence. The inhibitor studies were performed similarly, except the inhibitor was added 15 min before the addition of the PBS containing 1 μM calcein-AM.
Rhodamine 123 Efflux Assay.
Cells were cultured on 60-mm dishes under conditions above. First the cells were washed with PBS and then incubated in warm medium containing Rhodamine 123 (1 μg/ml). After a 1-h incubation, the cells were washed with ice-cold PBS and then resuspended into warmed drug-free medium for 1 h. Subsequently, the cells were isolated and intracellular rhodamine was determined by fluorescence-activated cell sorting.
Results
A mouse liver cDNA library was initially screened with a 1.1-kb fragment of the mouse SPGP cDNA (see Experimental Procedures) that was 78.5% identical with the pig SPGP (Childs et al., 1995). Subsequently, a combination of PCR and hybridization was used to isolate the full-length murine SPGP. The mouse SPGP cDNA is 4717 base pairs with the initiator methionine at position 89 agreeing best with Kozak rules (Kozak, 1989). To confirm that the cDNA had the entire SPGP open-reading frame, the cDNA was subcloned into pcDNA3 and in vitro translated. The in vitro translated transcript was size-fractionated on a polyacrylamide gel and, after autoradiography, estimated at 150 kDa (Fig. 1A). This value is remarkably close to the predicted mass of SPGP based on the 1321 amino acid open-reading frame (predicted mass, 146.7 kDa).

In vitro translation of the mouse SPGP cDNA and deduced amino acid sequence of the murine SPGP. A, the mouse SPGP cDNA was subcloned into pcDNA3 and then in vitro translated with the TNT coupled reticulocyte lysate system followed by an analysis on 7.5% denaturing polyacrylamide gel. Lane 1, empty pcDNA3 vector; lane 2, mouse SPGP cDNA cloned in pcDNA3, clone 7; lane 3, luciferase positive control running at approximately 61 kDa. B, comparison of the deduced amino acid sequence of the mouse SPGP (mus, top line) with the rat SPGP (rat, bottom line). Predicted TM domains (TM1–12) are underlined and the walker A and B motifs for ATP binding and the ABC signature motif are bold. The peptides used to generate the C219 and SPGP antibodies are bold, and the peptide used to generate mdr (Ab-1) IgG is underlined.
The predicted protein topology of SPGP reveals a protein with 12 transmembrane (TM)-spanning domains (Fig. 1B). In addition, the typical Walker A and B nucleotide binding motifs and the ABC family signature sequence. Further analysis reveals at least four candidate sites for N-linked glycosylation. The percentage amino acid identity of SPGP among ABC family members was 48% with human MDRs 1 and 3 and mouse mdr3, and 47% with mouse mdr1 and mdr2. Moreover, the identity of murine SPGP with human and rat SPGPs was about 80% and 90%, respectively, with the greatest divergence between mouse and rat SPGP found in the predicted TM domains (ranging from 81 to 89% similarity).
Although the human bile salt export pump (BSEP) gene localized to a region in chromosome 2q24-31 (Strautnieks et al., 1998), the imperfect synteny between mouse and human did not permit us to assume that murine BSEP was on chromosome 2. A murine BAC library was screened by hybridization with the 1.1-kb fragment of the mouse SPGP cDNA. Of the four BAC clones obtained, one clone hybridized with over 25 kb of genomic DNA after hybridization with the full-length SPGP cDNA. However, based on the estimated size of the murinemdr1 genes (approximately 60 kb), this is likely to be an underestimate because the 3′ and 5′ SPGP probes suggested that this BAC clone encompassed the entire murine SPGP gene. To confirm that this was indeed SPGP, this clone BAC1 was further characterized by sequence analysis for intron-exon borders using a combination of hybridization and PCR analysis (Fig. 2A). We used this approach to identify at least nine exons from both the 3′ and the 5′ portions of the BSEP gene. Interestingly, the exon sizes and location of the functional domains (e.g., Walker A and ABC signature) in the 3′ end of the gene (exons F, G, and H) are remarkably similar to that found for the murine mdr genes (Raymond and Gros, 1989) and suggest common ancestry between these genes. This BAC-containing SPGP was then labeled with digoxigenin dUTP, and FISH was performed as described in Experimental Procedures (Fig. 2B). Analysis of 80 metaphase spreads from mouse embryo fibroblast cells indicated that the mouse BSEP gene was located at an area that corresponds to band 2C1.3 (Fig. 2C). The labeling of chromosome 2 was confirmed by using a telomeric probe specific for chromosome 2. Thus, murine BSEP localizes to a region syntenic with human chromosome chromosome 2q24-31, a region that has been genetically linked to hypersusceptibility to cholesterol induced gallstones in mice (Lammert et al., 1997).

Diagram of the mouse SPGP gene organization and chromosomal localization. A, Nine exons were identified in the SPGP BAC, four exons labeled as exon A to E in the 5′ part of SPGP, and four exons labeled as F to I in the 3′ end in the gene. The DNA sequence coding for the TM1 was found in the exons D and E, whereas the sequence coding for the Walker A and the ABC signature in 3′ end were found in the exon F and H, respectively. B, chromosomal localization of the mouse SPGP gene by FISH analysis of hyperdiploid mouse embryo fibroblast metaphase chromosomes. Arrows indicate SPGP, which fluoresces green. C, ideogram indicating localization of murine SPGP.
To determine the pattern of tissue expression, we hybridized a mouse multitissue Northern blot with the 1.1-kb fragment of the mouse SPGP cDNA and detected a strong 5.5-kb mRNA signal in the liver only, not in the heart, brain, spleen, lung, kidney, testis, or skeletal muscle (Fig. 3A). We further confirmed the hepatic predominance of SPGP by analyzing the same tissues for SPGP protein by Western immunoblot; interestingly, we found very weak anti-SPGP immunoreactivity on immunoblots from total brain homogenate (data not shown). The same radiolabeled probe was used concurrently to hybridize a mouse embryo tissue Northern blot. This study revealed that, unlike the rat, murine SPGP is expressed in embryonic life and that expression, first detected at day E15, increased at day E17 (Fig.3B). However, despite an increase in SPGP expression in murine embryonic life, the amount of SPGP transcript was less abundant than adult liver. Thus, expression of SPGP in murine embryonic life, unlike rat, is probably caused by physiological differences between rat and mouse.
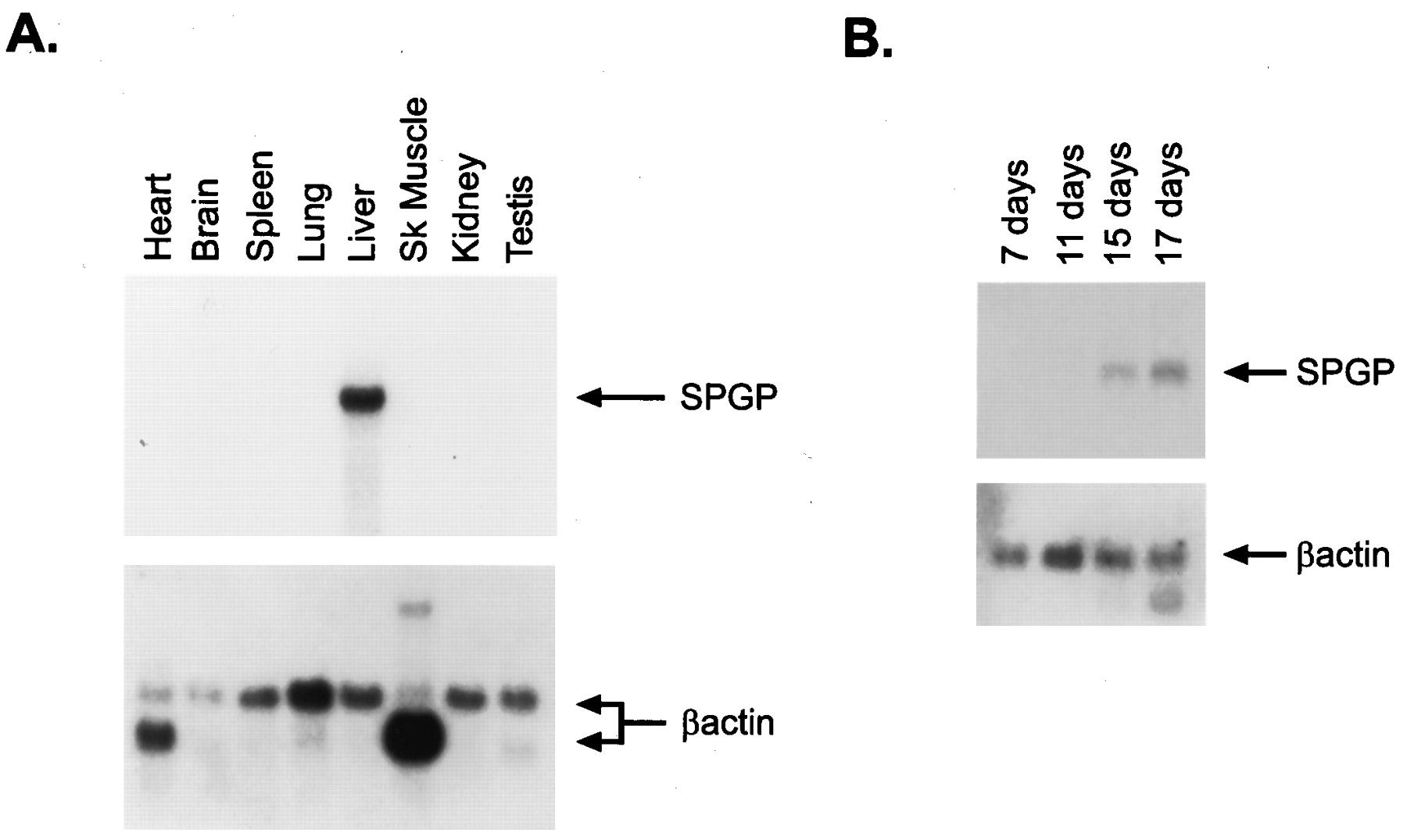
SPGP mRNA expression in developing mice and adult mouse tissue. A, Northern blot analysis of adult mouse RNAs using the 1.1-kb fragment of the mouse SPGP cDNA showed a strong overexpression of a 5.5-kb transcript in only the liver of adult mice. B, SPGP expression was detectable in the mouse embryo at 15 and 17 days. The blots were concurrently hybridized with the labeled SPGP cDNA to allow comparison of the level of SPGP expression.
We chose MDCKII and LLC-PK1 cell lines as hosts because derivatives of these cell lines, which stably express murine SPGPs closest homolog, human MDR1, were to be used for comparative transport studies. In addition, because membrane composition can affect Pgp function and substrate recognition (Sinicrope et al., 1992), and might similarly affect SPGP, we elected to express SPGP first in two different polarizing mammalian cells and compared its function. Finally, comparison of drug transport characteristics by SPGP and MDR1 would not be confounded by differences in cell background that could influence transport function. The murine SPGP cDNA, cloned into the MSCV-IRES-GFP vector (Fig. 4A), was transduced into MDCKII and LLC-PK1 cells (see Experimental Procedures). From a pool of GFP-positive cells, individual clones were screened by immunoblot for SPGP expression using an antibody to the carboxyl-terminal domain in SPGP (see Experimental Procedures). Specificity of the antiserum was tested by Western blot analysis against a bacterial fusion containing a carboxyl-terminal fragment of mouse SPGP and by studies performed below (seeExperimental Procedures). We also showed by Western blot that the SPGP protein is only detected in the membrane and not in the cytosol of the mouse liver (data not shown). MDCKII SPGP clones 2 and 5 (but not the MDCKII MDR1 or the MDCKII parent cell line) highly expressed SPGP (Fig. 4B). Two LLCPK SPGP cell lines derived from clones 2 and 7 were also characterized and found to express substantial amounts of SPGP with undetectable SPGP in the parent LLC-PK1 or in the LLCPK MDR1 cells overexpressing Pgp (Fig. 4B). Although the MDCKII SPGP comigrates with the liver SPGP, the SPGP expressed in LLC-PK1 migrates slightly faster. The difference in mobility is most likely caused by differences in the extent of its glycosylation in MDCKII and LLC-PK1 cells. Like rat SPGP (Childs et al., 1995, 1998), murine SPGP is also recognized by an antibody to this MDR1/Pgp epitope (Fig. 4B). To further demonstrate the specificity of our SPGP antisera, these same immunoblots were analyzed with polyclonal affinity-purified anti-mdr (Ab-1) peptide IgG (Calbiochem, San Diego, CA)—the MDR1 epitope overlaps the peptide used to generate the C219 monoclonal antibody (Childs et al., 1995) (see Fig. 1). These studies confirm that the SPGP antisera does not detect ectopically introduced MDR1 or mdr1a (data not shown) in either MDCKII or LLCPK cells; rather, the commercially available anti-mdr antibody recognizes both SPGP and MDR1. Further Northern blot analysis confirmed that SPGP mRNA was only detected in the cells that had been transduced with the SPGP retrovirus (Fig. 4C).
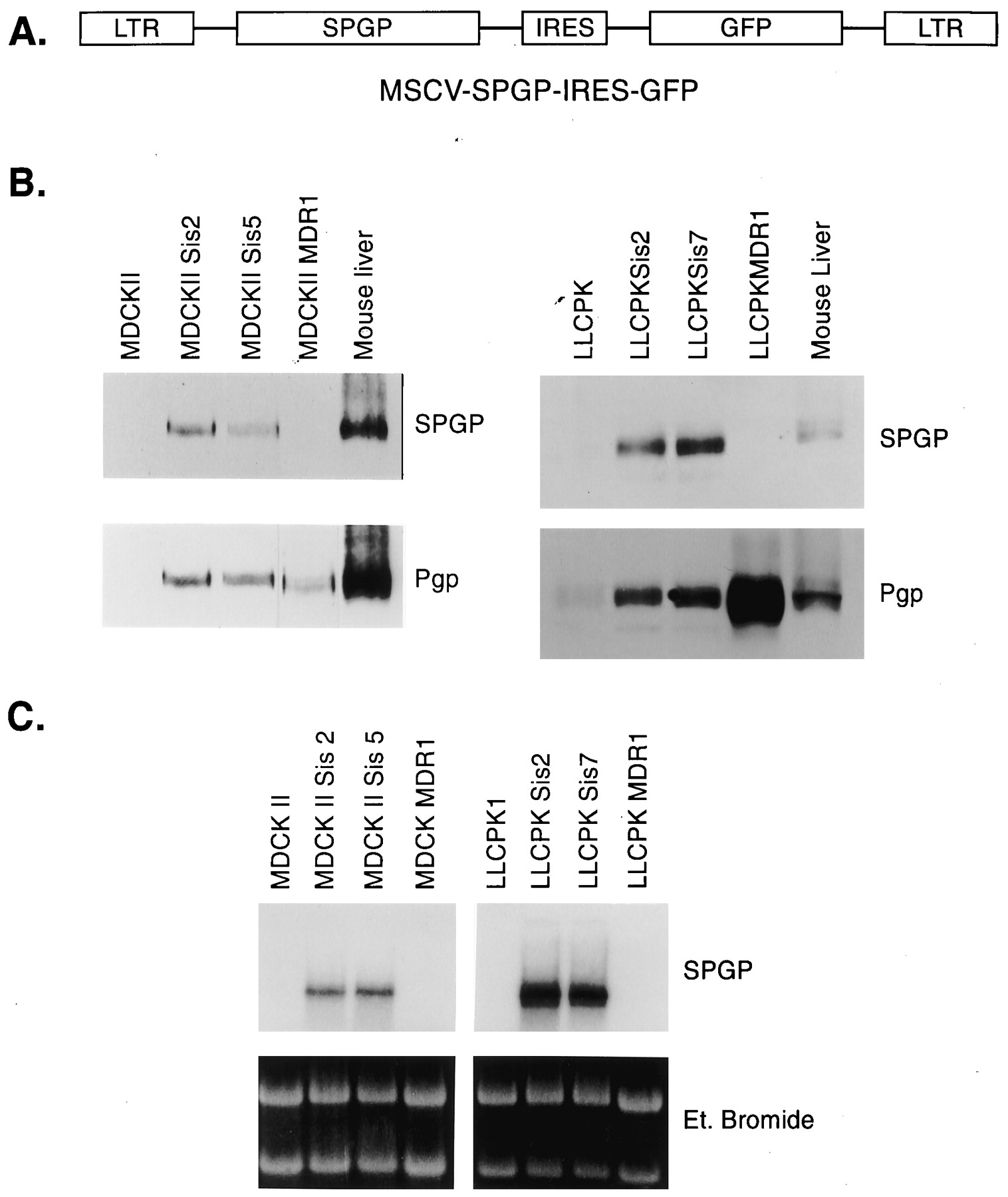
Analysis of the SPGP expression in MDCKII and LLC-PK1 transduced cell lines by western and northern blots. A, structure of the MSCV-SPGP-IRES-GFP vector used to transduce the 293T cells. B, protein (100 μg, total cell lysate) was loaded on a 7.5% denaturing polyacrylamide gel, transferred to a nitrocellulose filter, and developed with anti-SPGP IgG or mdr (Ab-1) (Calbiochem) to MDR1/Pgp. Mouse liver membranes were used as a positive control. C, ten micrograms of total RNA were resolved on a denaturing 1% agarose gel, stained with ethidium bromide, transferred to a nylon membrane, and hybridized with the 1.1-kb fragment of the mouse SPGP cDNA.
We initially evaluated SPGP function by measuring cellular accumulation of taurocholate in parental and SPGP derivative MDCKII and LLC-PK1 cells. MDCKII Sis2 cells accumulated lower levels of taurocholate than either MDCKII or MDCKII MDR1 cells (Fig.5A). These studies were then extended to the LLCPK1 cells incubated with identical concentrations of extracellular [3H]taurocholate. We found substantially lower levels of intracellular taurocholate in the SPGP-expressing cells (LLCPK Sis2) than in the parent cell line LLC-PK1 (Fig. 5B). It should be noted that cyclosporine A (CsA; 10 uM) increased taurocholate accumulation, but only in the LLCPK1 SPGP cells, not in the parental LLCPK1 cells (Fig. 5C). We do not have an explanation for the decreased taurocholate uptake in CsA-treated LLC-PK1 cells.
Some studies have suggested that the ABC transporters may have common substrate recognition sites (Ruetz et al., 1997) and, given the possibility that SPGP closely resembles MDR1, we assessed SPGP efflux function compared with MDR1 using vinblastine as a substrate. We first evaluated vinblastine uptake in the LLC-PK1, SPGP, and MDR1 cells (Fig.6A). SPGP cells accumulate substantially less vinblastine than the LLC-PK1 cells. In our next studies, we preloaded the cells with vinblastine (we used five times higher extracellular vinblastine for the MDR1 cells to achieve approximately 80% of the intracellular level found in the LLC-PK1 cells) and then assessed the efflux of the intracellular vinblastine. The rate of efflux from the LLC-PK1 was modest and the overexpression of SPGP accelerated the rate of vinblastine efflux; comparatively, however, the vinblastine efflux in SPGP cells was slower that in MDR1 cells (Fig.6B). Further studies assessed the comparative uptake kinetics of vinblastine in LLC-PK1, LLCPK MDR1, and LLCPK Sis7 cells (Table1). These studies revealed that the maximal rate of vinblastine uptake by MDR1 and SPGP cells was only 50% and 72%, respectively, of LLC-PK1 cells. Cumulatively, the uptake and efflux and kinetic studies revealed that vinblastine is a SPGP substrate.
Vinblastine uptake kinetics for SPGP cells compared with that of LLCPK MDR1 and LLC-PK1 cells
Because SPGP may be the transporter effluxing paclitaxel and digoxin in mdr1a/1b nullizygous animals ( Mayer et al., 1997; Schinkel et al., 1997a), we evaluated paclitaxel, digoxin, daunomycin, vincristine, and tamoxifen as potential SPGP substrates by examining drug uptake compared with the MDR1 cells (Fig. 7). With the exception of vinblastine, SPGP did not effectively transport the drugs tested. It should be noted that tamoxifen, although a reported MDRI inhibitor, seems unlikely to be a substrate for either MDR1 or SPGP. Further evidence that paclitaxel is not a SPGP substrate was demonstrated by the fact that [3H]paclitaxel efflux in the LLC-PK1 and SPGP cells was not different (data not shown). Further studies revealed that overexpression of SPGP did not confer paclitaxel resistance (data not shown), thus ruling out the possibility that SPGP overexpression cells might trap paclitaxel in an intracellular compartment and abrogate its cytotoxic effects. Cumulatively, these studies show that the murine SPGP does not transport paclitaxel.
To further evaluate SPGP transporter activity compared with MDR1, we assessed the accumulation of the fluorescent indicator and Pgp substrate calcein-AM (Tiberghien and Loor, 1996; Essodaigui et al., 1998) (Fig. 8). Calcein-AM is a nonfluorescent, hydrophobic compound transported by MDR1/Pgp and MRP1 (Essodaigui et al., 1998) that is hydrolyzed rapidly by cytoplasmic esterases to release the membrane-impermeable, fluorescent calcein moiety that is not a substrate for Pgp (Essodaigui et al., 1998). The rate of calcein-AM uptake into a population of cells can be measured by monitoring the rate of fluorescence increase. The rate of intracellular calcein accumulation in cells expressing MDR1 was considerably reduced compared with that of the parental cells LLC-PK1 (Fig. 8). More importantly, calcein fluorescence was reduced in both LLCPK SPGP cell lines compared with the LLC-PK1. Like vinblastine, the rate of calcein-AM uptake in LLCPK SPGP cell lines was intermediate between LLC-PK1 and LLCPK MDR1 cells, indicating that calcein-AM is a SPGP substrate. Further studies revealed that a well known Pgp substrate, rhodamine 123, was not effluxed by SPGP cells (Fig.9).

SPGP cells transport Calcein-AM. LLC-PK1, LLCPKSis7, LLCPKSis2, and LLCPK MDR1 cells were cultured in a Costar 96-well plate on day zero at 105 cells/well in medium. On day 1, medium was removed and the cells were incubated in 1 μM calcein-AM and calcein fluorescence monitored by spectrofluorometry. The plate was scanned at 3-min intervals repeated 11 times over 30 min at room temperature. The results are expressed as fold increase of Calcein fluorescence compared with the control (time zero). Each point is the mean ± S.E. of two independent experiments done in sextuplicate.

SPGP does not efflux Rhodamine 123, unlike MDR1/Pgp. LLC-PK1, LLCPKSis7, and LLCPK MDR1 cells were cultured on 60-mm plastic dishes. On day 1, medium was removed and the cells were incubated in rhodamine 123 at 1 μg/ml for 1 h followed by a washing with 1× PBS and then resuspension in fresh, drug-free medium for 1 h. This is a representative experiment of two independent experiments showing essentially identical results.
Because a number of inhibitors of MDR1/Pgp have been identified by the calcein-AM transport assay, we evaluated several of these as inhibitors of SPGP-mediated efflux of calcein-AM (Fig.10). We found that reserpine had no effect on SPGP efflux of calcein-AM, but readily inhibited MDR1. The MDR1/Pgp inhibitor CsA potently inhibited MDR1/Pgp efflux of calcein-AM. In contrast, CsA produced a small inhibition of SPGP. MDR1/Pgp and other ABC transporters can transport hydrophobic peptides—the best known example is the hydrophobic a-factor, a substrate for ste6p (Nijbroek and Michaelis, 1998). Moreover, because many peptide drugs are excreted unchanged from the liver via the bile, it seemed likely that SPGP might interact functionally with hydrophobic peptides. Therefore, we assessed whether a linear peptide, ditekiren, that had been previously demonstrated to be transported across hepatic canalicular membranes (Takahashi et al., 1997) was an inhibitor of SPGP. We found that ditekiren inhibited efflux of calcein-AM in SPGP cells. In contrast, although ditekiren inhibited MDR1 efflux of calcein-AM at 20 μM, it was ineffective in inhibiting MDR1 efflux of calcein-AM at lower concentrations. Moreover, structural modifications to ditekiren demonstrated that only one analog (7188) of the three available inhibited calcein-AM efflux, but 7188 was less effective than ditekiren. Cumulatively, these studies have identified calcein-AM as a substrate for SPGP and that the peptide ditekiren can preferentially inhibit SPGP.

The peptide ditekiren inhibits calcein-AM transport. LLC-PK1, LLCPKSis7, and LLCPK MDR1 cells were cultured in a Costar 96-well plate on day 0 at 105 cells/well in medium. On day 1, medium was removed and the cells were preincubated for 15 min in the presence of PBS or PBS containing CsA, reserpine (RES), ditekirin (DT), or the ditekiren analog 7188, each at 20 μM. Calcein-AM (1 μM) was then added and calcein fluorescence was monitored by spectrofluorometry. The plate was scanned at 3-min intervals repeated 11 times over 30 min at room temperature. The results are expressed as fold increase of Calcein fluorescence compared with the control (time zero). Each point is the mean ± S.D. of two independent experiments done in sextuplicate.
Discussion
Before these studies, it was unknown whether murine SPGP functioned as a drug transporter in mammalian cells. To determine SPGP's drug transport function, we isolated the murine SPGP, cloned it into a retroviral expression vector, and then transduced SPGP into LLCPK1 and MDCKII cells. These cells allowed us to perform comparative studies with one of its closest homologues, MDR1/Pgp. Our studies indicate that like MDR1, murine SPGP can transport vinblastine and calcein-AM. These studies also indicate that fundamental substrate differences exist between MDR1/Pgp and murine SPGP. For instance, SPGP is unable to transport paclitaxel, daunomycin, vincristine, digoxin, tamoxifen, and rhodamine 123. Furthermore, murine SPGP was incapable of inhibiting paclitaxel uptake or accelerating its efflux. In contrast, SPGP decreased accumulation and increased efflux of vinblastine. Additional studies revealed that the linear hexapeptide ditekiren inhibited calcein-AM transport by SPGP at low concentrations, whereas MDR1 was only effectively inhibited at higher concentrations of ditekiren. We also found that SPGP does not, unlike MDR1/Pgp, transport a variety of hydrophobic fluorescent probes (Homolya et al., 1993), because it is unable to efflux Rhodamine 123. Thus, these studies demonstrate inherent differences between SPGP and MDR1 in their ability to translocate drugs.
Using the murine SPGP cDNA, we isolated the SPGP gene and localized murine SPGP to chromosome 2 (Fig. 4). This chromosomal position is different than for murine Pgps that are positioned on chromosome 5 (Raymond et al., 1990) and supports the idea of evolutionary divergence between these genes. However, our partial determination of the murineSPGP gene structure suggests that SPGP and thePgp genes share a common ancestry. For instance, the size of several of the 3′-terminal exons are almost identical among the murine SPGP, and the murine, hamster, and human Pgps (Raymond and Gros, 1989). The slight differences in the exon size of these genes (1–3 base pairs) could be caused by a phenomenon referred to as “junction sliding,” which may also explain the larger open-reading frame for SPGP (Yonekura et al., 1988). Nevertheless, the positions of the different functional domains (e.g., Walker A and ABC signature) are almost identical within the exons of SPGP and the Pgps. Thus, although limited, our structural analysis of the murine SPGP gene supports the concept of a common ancestor between SPGP and the Pgps.
This investigation also revealed differences in temporal expression of the rat and murine SPGP genes. Murine SPGP is expressed late in embryonic development (day 15 of gestation; Fig. 3B). In contrast, expression of rat SPGP occurs postpartum (Childs et al., 1998). This species difference in ontogeny of SPGP mRNA is not caused by the inability of rat liver to accumulate bile acids, because both the Na+-dependent (Ntcp) and Na+-independent (Oatp1) bile acid transporter are expressed in the rat liver during embryonic development (Boyer et al., 1993; Dubuisson et al., 1996). An alternate explanation is anatomic, in that rats do not have a gallbladder to store bile acids. We speculate that the murine SPGP is expressed during embryogenesis because excreted bile acids can be stored in the gallbladder, unlike the rat, which lacks a gallbladder. An alternative explanation is that the profile of bile acids expressed in different species impacts SPGP expression. SPGP may be up-regulated in embryonic life because the bile acid composition differs between mouse and rat (Beher et al., 1969). For example, the bile acid chenodeoxycholate is a major constituent of bile in the rat, but is poorly represented in bile from mice (Beher et al., 1969). Thus, it is conceivable that bile acids may up- or down-regulate SPGP expression. This should be the subject of future investigation. It is interesting to note that human SPGP is expressed in embryonic life (J.D.S. and V.L., unpublished observations) and suggests that human SPGP may be regulated similarly to mouse SPGP.
Our results also suggest that inhibition of SPGP by drugs may be one mechanism of drug-induced cholestasis (Katagiri et al., 1992; Crocenzi et al., 1997). Compared with the host cell lines, we demonstrated that in both the MDCKII SPGP and LLCKPK SPGP ectopically overexpressing SPGP, the accumulation of the bile acid taurocholate was much lower and that CsA partially but selectively increased intracellular accumulation of taurocholate in SPGP cells. The role of CsA as an inhibitor of SPGP (albeit weak) is also supported by our findings that CsA inhibits SPGP transport of calcein-AM. These findings may provide a biological explanation for the fact that CsA induces cholestasis in some post-transplantation cases (Chan and Schaffer, 1997). These results and our findings suggest that CsA-induced cholestasis is due in part to the inhibition of SPGP activity.
There is considerable interest in identifying the alternative paclitaxel and digoxin hepatic transporters that have been revealed inmdr1a/1b knockout mice (Mayer et al., 1997; Schinkel et al., 1997a). Certainly the canalicular localization of SPGP (Gerloff et al., 1998) and the increased resistance to paclitaxel reported for cells ectopically expressing rat SPGP (Childs et al., 1998) implicated SPGP as the hepatic paclitaxel transporter in mice. However, this conclusion seems untenable in light of our results demonstrating that drug uptake of paclitaxel and digoxin are unaffected by SPGP. However, because paclitaxel is readily metabolized in the liver (at least 25% of a dose of paclitaxel is recovered in the bile as metabolite) and because a greater proportion of paclitaxel metabolites are found in the feces of the mdr1a(−/−) compared with mdr1a(+/+) mice (Sparreboom et al., 1997), it is possible that SPGP may transport paclitaxel metabolites. This may not be the case for digoxin because it is less readily metabolized; hence, it is likely that another transporter is involved in digoxin efflux.
Of particular interest were the differences in drug substrates and inhibitors between SPGP and MDR1. An obvious basis for functional differences between SPGP and Pgp is the amino acid variation in the TM domains. The TM α helices of the Pgp molecules are important domains in determining substrate recognition and binding (Taguchi et al., 1997); however, other “domains” are undoubtedly important (Safa et al., 1990). The lower rates of both calcein-AM and vinblastine efflux support the idea that differences in substrate binding exist; however, to definitively prove this, further studies are needed. Our studies also suggest that the hexapeptide ditekiren interacts differently with SPGP and Pgp. Another explanation for the less effective ditekiren inhibition of MDR1 may be that there is a greater amount of MDR1 than SPGP protein. Although this is suggested by the level of immunoreactivity with the mdr1 (Ab-1) antibody (Fig. 4B), it is possible that the two transporters are expressed to equivalent levels in the derivative cell lines and the mdr1 antibody (Ab-1) has different affinity for the SPGP compared with MDR1 epitope. Several members of the ABC transporter superfamily interact with hydrophobic peptides (the first identified being ste6p, the ABC transporter that transports the hydrophobic a-factor mating peptide). It should also be noted that MDR1 has been reported to transport hydrophobic peptides (Sarkadi et al., 1994). Thus, it is conceivable that SPGP may transport peptides; this concept is intriguing because many peptide drugs are found unmetabolized in bile. Finally, given that one of the mutations in human SPGP leads to an apparent loss of bile acid transport ability (Strautnieks et al., 1998), it would be intriguing to determine whether a similar mutation in murine SPGP affects its drug transport function.
The tissue distribution of SPGP is also of interest. Our results demonstrate that SPGP was expressed primarily in the livers of normal mice. Although SPGP is reportedly expressed in rat liver tumors (Childs et al., 1998), the expression of SPGP in primary or secondary human liver tumors is unknown. Because the vinca alkaloid vinblastine is a substrate for SPGP, knowledge of SPGP expression in human liver tumors would provide therapeutic insight into the utility of using this chemotherapeutic and the potential role SPGP may have in the responsiveness to cancer chemotherapy. Although initial reports found that SPGP expression was restricted to liver, a recent PCR-based study suggests that this gene may be expressed extrahepatically (Torok et al., 1999). Our own immunoblot analysis of total mouse brain homogenates indicates weak expression of murine SPGP. Weak expression of SPGP in the total brain isolate may be attributable to selective expression of SPGP in only a finite number of cells in the brain, such as the epithelial cells lining the blood brain barrier, like Pgp (Schinkel et al., 1997b), or in the epithelial cells of the choroid plexus, like MRP1 (Rao et al., 1999). Additional analysis of SPGP expression in the brain should reveal whether brain SPGP serves as an additional barrier to the penetration of drugs into the central nervous system. The expression of SPGP in the brain would be intriguing because recent studies have functionally described a bile acid (taurocholate) transporter in the blood-brain barrier that effluxes taurocholate and interacts with peptides (Kitazawa et al., 1998).
Much work still remains to done using the SPGP model cell system to define the pharmacological profile (substrate and/or inhibitor) of drugs that interact with SPGP; these cells will serve as a model to screen for drugs and steroids that may cause untoward pathophysiological effects (e.g., cholestasis) because of interactions with SPGP.
Acknowledgments
We thank the members of the St. Jude Children's Research Hospital Hartwell Center for Biotechnology for their assistance in DNA sequencing and peptide synthesis. We gratefully acknowledge the support of Dr. A. W. Nienhuis during the course of these studies.
Footnotes
-
Send reprint requests to: Dr. John D. Schuetz, Department of Pharmaceutical Sciences, St. Jude Children's Research Hospital, 332 N. Lauderdale Ave., Memphis, TN 38105. E-mail:john.schuetz{at}stjude.org
-
This work was supported by National Institutes of Health Grants ES/GM 5851, ES/GM 8568, CA 21765, CA 23099, and GM 31304, and by the American Lebanese Syrian Associated Charities.
- Abbreviations:
- Pgp
- P-glycoprotein
- SPGP
- sister of P-glycoprotein
- ABC
- ATP-binding cassette
- MDR
- multidrug resistance
- AM
- acetoxymethyl ester
- kb
- kilobase
- EST
- expressed sequence tag
- BAC
- bacterial artificial chromosome
- PCR
- polymerase chain reaction
- FISH
- fluorescence in situ hybridization
- GST
- glutathione transferase
- GFP
- green fluorescence protein
- BSEP
- bile salt export pump
- CsA
- cyclosporine A
- TM
- transmembrane
- Received July 6, 1999.
- Accepted September 23, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}