


Abstract
Anandamide acts as a full vanilloid receptor agonist in many bioassay systems, but it is a weak activator of primary afferents in the airways. To address this discrepancy, we compared the effect of different vanilloid receptor agonists in isolated airways and mesenteric arteries of guinea pig using preparations containing different phenotypes of the capsaicin-sensitive sensory nerve. We found that anandamide is a powerful vasodilator of mesenteric arteries but a weak constrictor of main bronchi. These effects of anandamide are mediated by vanilloid receptors on primary afferents and do not involve cannabinoid receptors. Anandamide also contracts isolated lung strips, an effect caused by the hydrolysis of anandamide and subsequent formation of cyclooxygenase products. Although capsaicin is equally potent in bronchi and mesenteric arteries, anandamide, resiniferatoxin, and particularly olvanil are significantly less potent in bronchi. Competition experiments with the vanilloid receptor antagonist capsazepine did not provide evidence of vanilloid receptor heterogeneity. Arachidonoyl-5-methoxytryptamine (VDM13), an inhibitor of the anandamide membrane transporter, attenuates responses to olvanil and anandamide, but not capsaicin and resiniferatoxin, in mesenteric arteries. VDM13 did not affect responses to these agonists in bronchi, suggesting that the anandamide membrane transporter is absent in this phenotype of the sensory nerve. Computer simulations using an operational model of agonism were consistent, with differences in intrinsic efficacy and receptor content being responsible for the remaining differences in agonist potency between the tissues. This study describes differences between vanilloid receptor agonists regarding tissue selectivity and provides a conceptual framework for developing tissue-selective vanilloid receptor agonists devoid of bronchoconstrictor activity.
Capsaicin, the hot ingredient in chili peppers, activates a subpopulation of primary sensory neurons, generally referred to as polymodal nociceptors (Holzer, 1991; Julius and Basbaum, 2001). These nociceptive nerves participate not only in afferent signaling, but also in local responses to potentially harmful stimuli via the release of neurotransmitters from their peripheral nerve endings (Maggi, 1991; Holzer, 1992). The receptor for capsaicin, termed the vanilloid receptor (Szallasi and Blumberg, 1999) or TRPV1 (Montell et al., 2002), is a heat- and proton-gated cation channel, which was originally proposed bySzolcsanyi and Jancso-Gabor (1975) and later cloned by Julius and coworkers (Caterina et al., 1997). Both native and artificially expressed vanilloid receptors are activated by the endogenous lipid anandamide (Zygmunt et al., 1999; Smart et al., 2000). Anandamide was originally isolated from porcine brain as the first endogenous cannabinoid (Devane et al., 1992), and its biosynthesis has subsequently been demonstrated in neurons (Di Marzo et al., 1994), macrophages (Di Marzo et al., 1996), and recently in lung (Calignano et al., 2000). In contrast to capsaicin, anandamide and the synthetic vanilloids and analgesic agents olvanil and SDZ 249–665 do not induce airway obstruction in vivo (Wrigglesworth et al., 1996; Stengel et al., 1998; Calignano et al., 2000; Urban et al., 2000). Consistent with these observations, recent studies suggest that anandamide is a weak constrictor of isolated airways compared with capsaicin (Craib et al., 2001; Tucker et al., 2001). This is somewhat surprising, because both anandamide and olvanil are effective vanilloid receptor agonists in many other bioassay systems (Hughes et al., 1992; Szallasi and Blumberg, 1999; Zygmunt et al., 1999; Smart et al., 2000; Ralevic et al., 2001).
In the present study, we examined the effects of different vanilloid receptor agonists in guinea pig airways and mesenteric arteries. Both of these tissues are richly innervated with capsaicin-sensitive sensory nerves, but the neurotransmitter systems responsible for the physiological readouts of nerve activation are different (Fujimori et al., 1990; Jansen et al., 1990; Franco-Cereceda, 1991; White et al., 1993; Lundberg, 1995; Zygmunt et al., 1999). We show that these two phenotypes of primary sensory nerve respond differently to the nonpungent N-acyl amines anandamide and olvanil.
Materials and Methods
Tension Recordings.
Male Dunkin-Hartley guinea pigs (300–500 g) were killed by cervical dislocation followed by exsanguination. Mesenteric arteries and main bronchi were removed and divided into ring preparations, which were 1 to 2 mm long (arterial segments) or 1 to 2 rings of cartilage wide (main bronchi). Strips of lung (approximately 3 × 3 × 6 mm) were cut longitudinally from parenchyma distal to the major airways. The tissue preparations were mounted between two metal pins in organ baths (5 ml) containing warmed (37°C) physiological salt solution of the following composition: 119 mM NaCl, 15 mM NaHCO3, 4.6 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 1.5 mM CaCl2, and 6.0 mM (+)-glucose. The physiological salt solution was continuously bubbled with a mixture of 95% O2 and 5% CO2, resulting in a pH of 7.4. During an equilibration period of approximately 1 h, the preparations were repeatedly stretched until a passive tension of approximately 2 to 4 mN (arterial segment), 3 to 4 mN (bronchial ring), or 2 mN (parenchymal strip) was obtained. Isometric tension was measured as described previously (Högestätt et al., 1983). Experiments with vascular preparations were carried out in the presence of indomethacin (10 μM) andN ω-nitro-l-arginine (300 μM). Main bronchi were studied in the presence of indomethacin (10 μM),N ω-nitro-l-arginine (300 μM), thiorphan (10 μM), enalaprilate (10 μM), and timolol (10 μM) to inhibit cyclooxygenase, nitric oxide synthase, neutral endopeptidase, angiotensin-converting enzyme, and β-adrenoceptors, respectively. The incubation time with all drugs was at least 30 min, and each preparation was exposed to only one treatment.
To assess the contractile capacity, mesenteric arteries, main bronchi, and lung strips were initially contracted by U46619 (100 nM), carbachol (10 μM), and histamine (10 μM), respectively. Relaxant responses in mesenteric arteries were expressed as the percentage of reversal of a contraction elicited by U46619 (100 nM). Contractions in main bronchi and lung strips were expressed as the percentage of the initial contractile response to carbachol and histamine, respectively. Agonists were added cumulatively to determine concentration-response relationships. The −10log of the agonist concentration eliciting half-maximal responses (pEC50) was determined by nonlinear regression (Prism 3.0; GraphPad Software Inc., San Diego, CA). Emax refers to the maximal response achieved. Data are expressed as mean ± S.E.M. Statistical analyses were performed by use of the Student's t test or analysis of variance followed by Bonferroni's post hoc test (Prism 3.0). Statistical significance was accepted when P < 0.05.
Computer Simulations.
An operational two-step model was created from a combination of logistic functions as described by the operational model of agonism (Black and Leff, 1983). According to the original model (Black and Leff, 1983), the first step represents the binding of the agonist to its receptor, and the second step represents the effector system. Although the present bioassay systems are very complex, comprising vanilloid receptor activation, neurotransmitter release, postjunctional receptor activation, and muscle contraction or relaxation, it may still be described by a two-step model provided that a slope coefficient is introduced in the first equation. Thus, the first step represents binding of the agonist to the vanilloid receptor and neural activation (eq. 1), whereas the second step represents all subsequent events in the signal pathway (eq. 2).
Drugs.
The following drugs were used in the course of the study: anandamide (Cayman Chemical, Ann Arbor, MI); arachidonic acid, carbachol, calcitonin gene-related peptide 8–37, histamine,N ω-nitro-l-arginine, phenylephrine, phenylmethylsulfonyl fluoride, resiniferatoxin, thiorphan, timolol, and WIN 55212-2 (Sigma Chemical, St Louis, MO); arachidonoyl-5-methoxytryptamine (Synthelec, Lund, Sweden); capsaicin, capsazepine, HU-210, olvanil (Tocris Cooksin Inc., Bristol, UK); enalaprilate (Merck-Sharp & Dohme, Stockholm, Sweden); indomethacin (Confortid; Dumex, Copenhagen, Denmark); SR 141716A, SR 48,968, SR 140,333 (SANOFI Research Center, Montpellier, France); andU46619 (Upjohn, Kalamazoo, MI).
Results
Functional Responses to Capsaicin in Main Bronchi and Mesenteric Arteries.
In main bronchi, capsaicin evoked concentration-dependent contractions (Fig.1). Substance P and neurokinin A also contracted this preparation (Fig. 1). The calcitonin gene-related peptide (CGRP) receptor antagonist CGRP 8–37 (3 μM) and the selective NK1 receptor antagonist SR 140,333 (100 nM) did not inhibit the capsaicin-induced contractions. The selective NK2 receptor antagonist SR 48,968 (100 nM) caused a small inhibition of these contractions and abolished them when combined with SR 140,333.

Capsaicin induces concentration-dependent contractions in main bronchi (A) and relaxations in mesenteric arteries (B) of guinea pig. A, in main bronchi, contractions induced by capsaicin were partially inhibited by the NK2 receptor inhibitor SR 48,968 (100 nM) and abolished in the additional presence of the NK1 receptor inhibitor SR 140,333 (100 nM), which alone was without effect (n = 4). B, in mesenteric arteries, the CGRP receptor antagonist CGRP 8–37 (3 μM) inhibited relaxations to capsaicin, whereas a combination of SR 140,333 (100 nM) and SR 48,968 (100 nM) was without effect (n = 5). Traces show typical responses to capsaicin in main bronchi at resting tension and mesenteric arteries contracted by U46619. C, concentration-response curves for the contraction elicited by substance P (SP) and neurokinin A (NKA) in main bronchi (n = 4), and the relaxation evoked by CGRP in mesenteric arteries (n = 6). The slope coefficient was larger for CGRP (6.6) than for SP (1.3; P < 0.001) and NKA (1.1; P < 0.001). Data are expressed as mean ± S.E.M.
Capsaicin and CGRP completely relaxed guinea pig mesenteric arteries contracted with 100 nM U46619 (Fig. 1). CGRP 8–37 (3 μM) almost abolished the capsaicin-induced relaxations, whereas a combination of SR 140,333 (100 nM) and SR 48,968 (100 nM) had no effect (Fig. 1).
Functional Responses to Anandamide in Main Bronchi and Mesenteric Arteries.
In main bronchi, anandamide (10 μM) produced small contractions that were not significantly enhanced by phenylmethylsulfonyl fluoride (PMSF; 100 μM), an inhibitor of fatty acid amidohydrolase (Fig. 2). The CB1 receptor antagonist SR 141716A (300 nM) did not affect the anandamide-induced contractions in the absence (n = 4, data not shown) or presence of PMSF (Fig. 2). However, when segments of main bronchi were incubated with the competitive vanilloid receptor antagonist capsazepine (3 μM), anandamide failed to elicit any contraction (Fig. 2). The CB1 and cannabinoid receptor subtype 2 receptor agonists HU-210 (1 μM) and WIN 55212-2 (1 μM) did not elicit a contraction or inhibit the response to anandamide (n = 3; Fig. 2). Furthermore, anandamide was unable to induce a relaxation of preparations submaximally constricted with carbachol whether or not the tissue was pretreated with capsaicin (n =2; data not shown).

Anandamide contracts main bronchi and relaxes mesenteric arteries via activation of vanilloid receptors. A, the vanilloid receptor antagonist capsazepine (300 nM), but not the CB1 receptor antagonist SR141716A (3 μM), inhibited contractions induced by anandamide in main bronchi (n = 4–5). The effects of capsazepine and SR141716A were studied in the presence of the FAAH inhibitor PMSF (100 μM), which by itself did not significantly potentiate contractions to anandamide (n = 6). B, capsazepine (300 μM) caused a rightward shift of the anandamide concentration-response curve in mesenteric arteries (n =6). Traces show the inability of the cannabinoid receptor agonists HU-210 and WIN 55212-2 to mimic the action of anandamide in main bronchi (A) and mesenteric arteries (B). Data are expressed as mean ± S.E.M. (*P < 0.05 compared with PMSF alone).
In mesenteric arteries, anandamide evoked concentration-dependent and complete relaxations, which were inhibited by capsazepine (300 nM; Fig.2). HU-210 (1 μM) and WIN 55212-2 (1 μM) failed to relax preparations that subsequently responded to anandamide (Fig. 2).
Functional Responses to Anandamide in Strips of Lung Parenchyma.
In strips of lung parenchyma, anandamide at a concentration of 100 μM produced robust contractions that were unaffected by SR141716A (300 nM) but were strongly inhibited by either PMSF (100 μM) or indomethacin (10 μM; Fig.3). Lower concentrations of anandamide (≤10 μM) failed to induce a contractile response. Arachidonic acid (10 μM) also induced contractions that were inhibited by 10 μM indomethacin (control: 84 ± 15%, n = 4; indomethacin: 18 ± 2%, n = 3). Capsaicin (10 μM), HU-210 (1 μM), and WIN 55212-2 (1 μM) did not contract lung parenchymal strips (n = 2–3; Fig. 3).
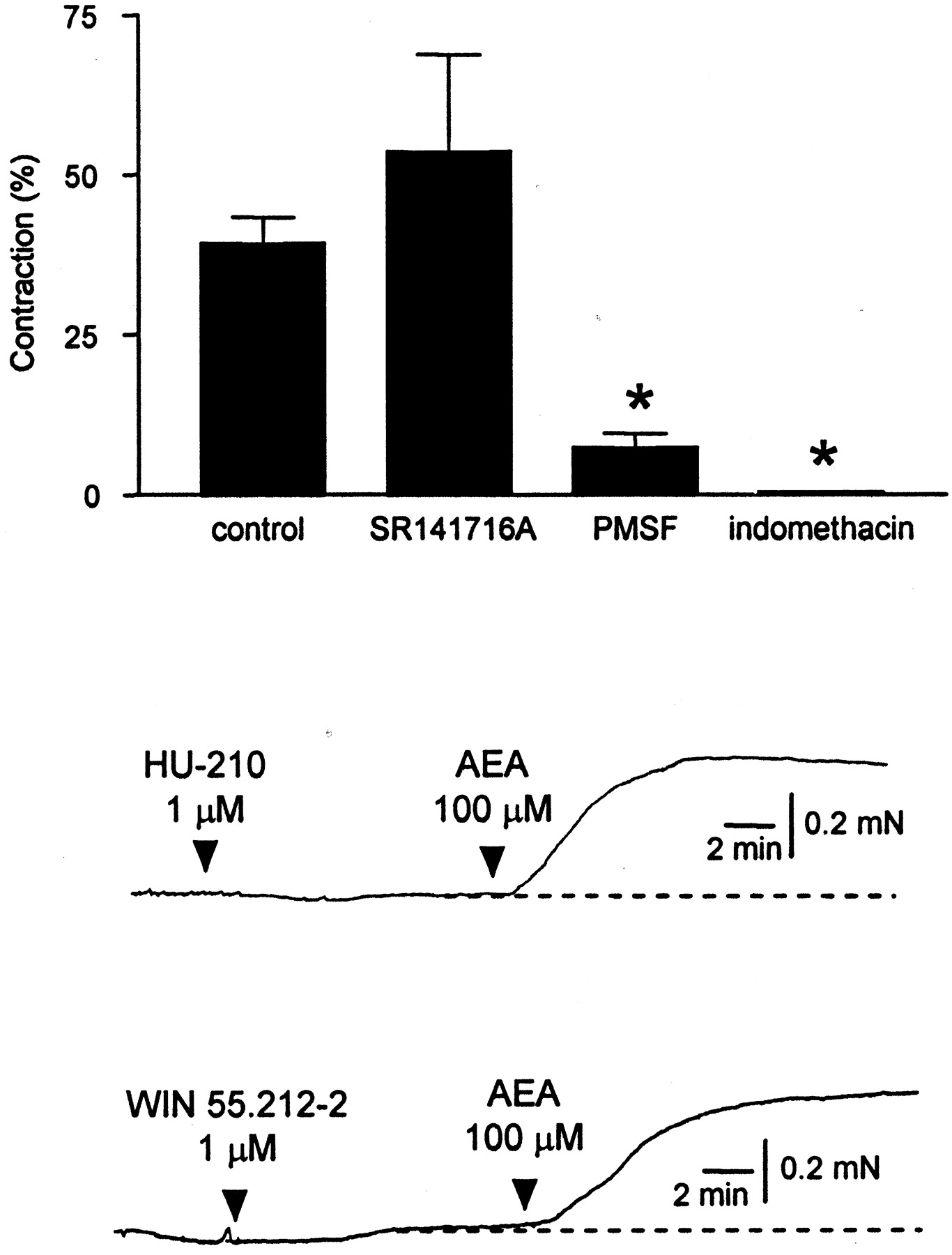
Anandamide (100 μM) contracts lung parenchymal strips of guinea pig. The anandamide-induced contractions were not inhibited by SR141716A (300 nM), whereas PMSF (100 μM) strongly inhibited and indomethacin (10 μM) abolished these contractions (n = 4). Traces show that the cannabinoid receptor agonists HU-210 and WIN 55212-2 do not contract lung strips subsequently responding to anandamide. Data are expressed as mean ± S.E.M. (*P < 0.05 compared with control).
Functional Responses to Vanilloid Receptor Agonists.
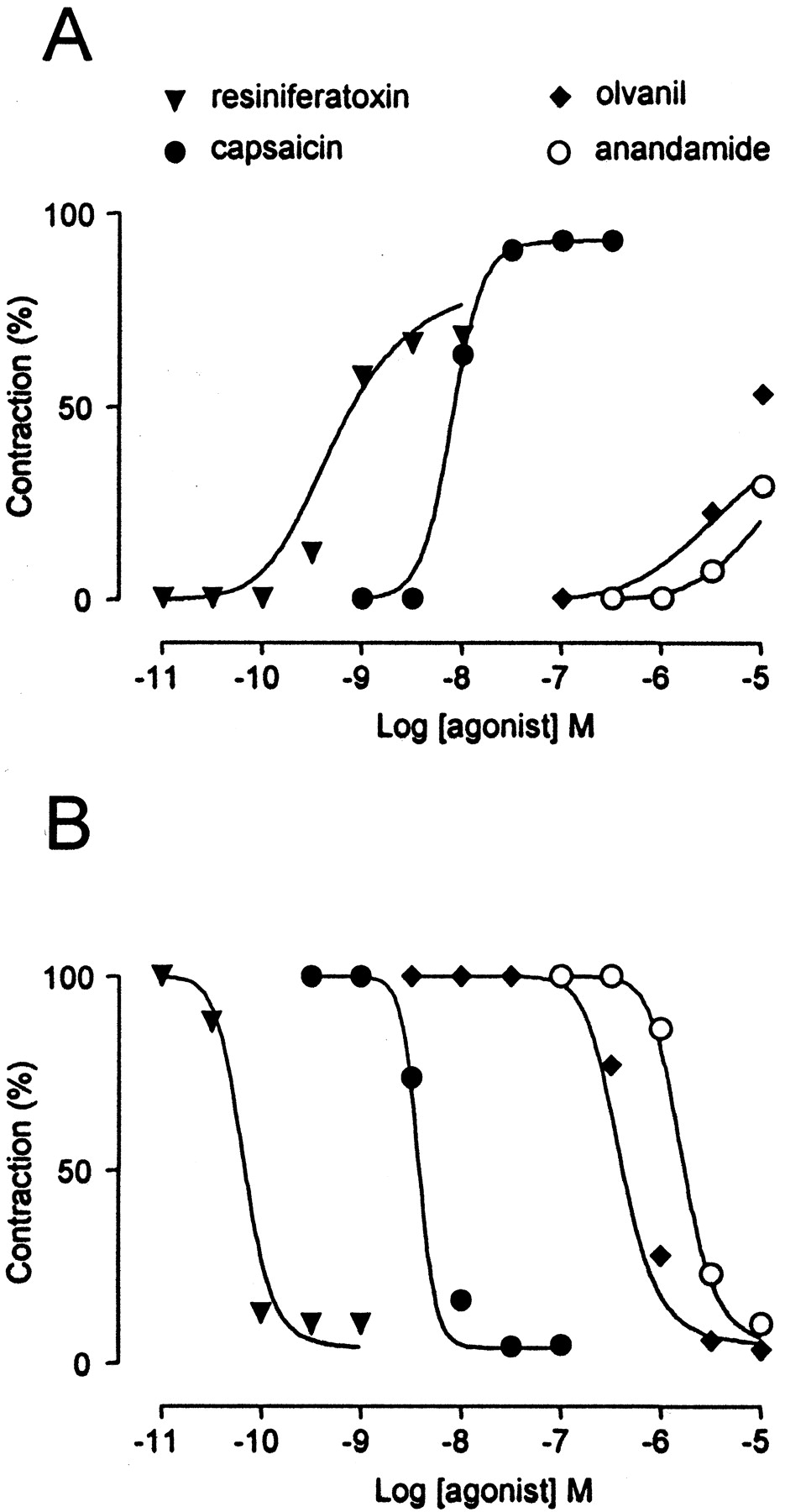
We next compared the effects of anandamide with three other vanilloid receptor agonists (capsaicin, resiniferatoxin, and olvanil) in main bronchi and mesenteric arteries. Whereas capsaicin was equally potent in both tissues, anandamide, resiniferatoxin, and particularly olvanil were less effective in main bronchi than in mesenteric arteries (Fig.4; Table 1). Anandamide only contracted main bronchi at the highest concentration tested (10 μM), precluding the calculation of EC50.

Concentration-response curves for the vanilloid receptor agonists resiniferatoxin, capsaicin, olvanil, and anandamide in main bronchi (n = 5, 7, 9, and 6, respectively) (A) and mesenteric arteries (n = 7, 6, 6, and 15, respectively) (B). Capsaicin was equally potent in the two tissues, whereas the other agonists were less potent in main bronchi than in mesenteric arteries. C, when responses to capsaicin were sampled at narrow intervals, the slope coefficient of the concentration-response curves was larger in arterial (19; n = 6) than in bronchial (8; n = 5) preparations (P< 0.05). Data are expressed as mean ± S.E.M.
Effects of vanilloid receptor agonists in guinea pig mesenteric arteries (MA) and main bronchi (MB)
The vanilloid receptor agonists had very steep concentration-response curves in both tissues with Hill coefficients well exceeding 1, consistent with a cooperative action on the vanilloid receptor (Szallasi and Blumberg, 1999). When responses to capsaicin were sampled at narrow concentration intervals (1/8 log units), the slope of the concentration-response curves was steeper in mesenteric arteries (slope coefficient = 19) than in main bronchi (slope coefficient = 8; Fig. 4).
Effect of the Vanilloid Receptor Antagonist Capsazepine.
To further characterize the receptor interactions, we constructed Schild plots for capsazepine using capsaicin and resiniferatoxin as agonists in both main bronchi and mesenteric arteries (Fig.5). Because the potencies of anandamide and olvanil were very low in main bronchi, competition experiments with these agonists could not be conducted in this tissue. All Schild plots had slope coefficients greater than unity (Table2). Schild plots with capsaicin and resiniferatoxin as agonists yielded similar pA2values in main bronchi and mesenteric arteries. The corresponding values obtained with olvanil (pA2) and anandamide (pA2′) in mesenteric arteries also did not differ from the pA2 values obtained with capsaicin and resiniferatoxin (Table 2).

Shild plots for capsazepine in main bronchi and mesenteric arteries using capsaicin (A),resiniferatoxin (B), and olvanil (C) as agonists (n = 5–7). In main bronchi, the potency of olvanil was too low to allow competition experiments with capsazepine. Data are expressed as mean ± S.E.M.
pA2 values and slope coefficients obtained from Schild plots for capsazepine
Effect of the Anandamide Membrane Transport Inhibitor Arachidonoyl-5-Methoxytryptamine.
A high-affinity uptake of anandamide is present in many cell types and represents a main pathway for the cellular uptake of fatty acid amides in neurons (Di Marzo et al., 1994; Beltramo et al., 1997). Di Marzo and coworkers recently described two selective inhibitors of this uptake mechanism (De Petrocellis et al., 2000; De Petrocellis et al., 2001a). We tested the effect of arachidonoyl-5-methoxytryptamine (VDM13), which, at a concentration of 50 μM, abolished the anandamide uptake in HEK293 cells exposed to 3.6 μM of anandamide (De Petrocellis et al., 2001a). Preincubation for 5 min with VDM13 (50 μM) caused a rightward shift of the concentration-response curve for olvanil (173-fold) and anandamide (2.3-fold) in mesenteric arteries (Fig.6; Table 3). VDM13 did not affect constrictor responses to olvanil and anandamide in main bronchi or the effects of capsaicin and resiniferatoxin in either tissue (Table 3). Although VDM13 reduced the difference in potency between the tissues, olvanil, anandamide, and resiniferatoxin were still more potent in mesenteric arteries than in main bronchi.

Concentration-response curves for the vanilloid receptor agonists anandamide (A) and olvanil (B) in the absence and presence of the anandamide membrane transport inhibitor VDM13 (50 μM) in mesenteric arteries (n = 5–7). Data are expressed as mean ± S.E.M.
Effects of VDM13 (50 μM) on responses to vanilloid receptor agonists in guinea pig mesenteric arteries (MA) and main bronchi (MB)
Computer Simulations.
According to classic receptor theory, the potency of an agonist is dependent on the amount of receptors or the receptor reserve (Black and Leff, 1983). Initial computer simulations using an operational two-step model of agonism indicated that the intrinsic efficacy of the agonist influences this relationship. Using data from experiments performed in the presence of VDM13 and allowing for differences in intrinsic efficacy between agonists, this model was used to construct concentration-response curves for the agonists (Fig. 7). The computer simulations provided a fit that was not significantly different from the original data (χ2 analysis = 64; df = 56). This fit was obtained with a 1.7-fold higher intrinsic efficacy (S m) for each agonist in mesenteric arteries than in main bronchi, consistent with a larger number of receptors (or receptor reserve) in the former than in the latter tissue. The intrinsic efficacy of capsaicin was estimated to be 14-, 13-, and 11-fold larger than that of olvanil, anandamide, and resiniferatoxin, respectively. Almost identical slope coefficients of the first system were estimated for the arterial (0.52) and bronchial (0.49) assays, consistent with activation of similar pathways in the two types of neurons. A higher slope coefficient of the second system was estimated in the former (9.9) than in the latter (6.1) assay, probably reflecting a higher slope coefficient for CGRP (6.6) in mesenteric arteries than for substance P (1.3) and neurokinin A (1.1) in main bronchi (p < 0.001). This is in line with the slope coefficient for capsaicin being larger in mesenteric arteries than in main bronchi.

Computer simulations of concentration-response curves for resiniferatoxin, capsaicin, olvanil, and anandamide in main bronchi (A) and mesenteric arteries (B) using an operational two-step model of agonism. The curves are superimposed on the experimental data, which were all obtained from experiments performed in the presence of the anandamide membrane transport inhibitor arachidonoyl-5-methoxytryptamine. The pK awas set to 9.89, 5.76, 6.58, and 5.78 for resiniferatoxin, capsaicin, olvanil, and anandamide, respectively, andK E was set to 1 in both tissues.E m was set to 92% in main bronchi and 4% in mesenteric arteries. In main bronchi, S mwas estimated to be 2.42, 25.6, 1.80, and 1.99 for resiniferatoxin, capsaicin, olvanil, and anandamide, respectively. These values were 1.72-fold larger for each agonist in mesenteric arteries.N S was estimated to be 0.49 and 0.52, andN E was estimated to be 6.13 and 9.86 for main bronchi and mesenteric arteries, respectively.
Discussion
Activation of primary sensory nerves by capsaicin in guinea pig main bronchi and mesenteric arteries leads to opposite functional responses that are mediated by different neurotransmitter systems. In accord with those from previous studies, our findings show that capsaicin-induced responses are mediated by NK1 and NK2 receptors in main bronchi (Ellis and Undem, 1994; Girard et al., 1997) and by CGRP receptors in mesenteric arteries (Fujimori et al., 1990; Zygmunt et al., 1999). Because vanilloid receptor–active drugs may behave differently in these test systems, it was of interest to compare the effects of anandamide with those of other vanilloid receptor agonists in these tissues. Anandamide, capsaicin, resiniferatoxin, and olvanil are all effective vasodilators of mesenteric arteries, whereas the synthetic cannabinoid receptor agonists HU-210 and WIN 55212-2 have no effect. Analysis of the Schild plots for capsazepine indicates that capsaicin, resiniferatoxin, and olvanil are all acting on the same population of vanilloid receptors. Capsazepine also inhibits the anandamide-induced relaxation, and the pA2′ value (6.9) agrees with the pA2 values obtained with the other vanilloid receptor ligands (6.8–7.2).
Surprisingly, olvanil and anandamide are weak agonists in main bronchi, whereas both of these compounds are potent and full agonists in mesenteric arteries. Although small, the responses to anandamide and olvanil in main bronchi are mediated by vanilloid receptors because they are abolished by capsazepine. The selective CB1 receptor antagonist SR 141716A as well as HU-210 and WIN 55212-2 do not affect the anandamide-induced contraction. It is therefore unlikely that anandamide activates inhibitory cannabinoid receptors on sensory nerves that would oppose any vanilloid receptor-mediated release of sensory neuropeptides, as has been demonstrated in rat skin and spinal cord (Richardson et al., 1998a,b). A rapid enzymatic degradation or metabolism via FAAH or cyclooxygenase cannot explain the weak effect of anandamide in main bronchi, because PMSF fails to significantly increase of the anandamide response and indomethacin was present in all experiments. Similar findings regarding the action of anandamide in guinea pig bronchus were recently reported (Craib et al., 2001; Tucker et al., 2001).
We further investigated the action of anandamide in isolated lung strips, because it has been proposed that anandamide induces a CB1 receptor-dependent contraction in this preparation (Calignano et al., 2000). We also found that anandamide at a high concentration is able to induce a contraction in lung strips, but its action does not involve cannabinoid receptors, because the responses are unaffected by SR 141716A. Vanilloid receptors also do not play a role in the anandamide-induced contraction, because capsazepine is unable to prevent the response. Furthermore, we found that HU-210, WIN 55212-2, and capsaicin are all lacking contractile activity, indicating that cannabinoid and vanilloid receptors do not mediate contraction in strips of lung parenchyma. However, inhibitors of FAAH and cyclooxygenase prevent the contractile response to anandamide, which is consistent with the hydrolysis of anandamide via FAAH to arachidonic acid and subsequent cyclooxygenase-dependent formation of bronchoconstrictor eicosanoids. Indeed, we found that arachidonic acid is a powerful contractile agent, acting through an indomethacin-sensitive pathway in this preparation. Such a metabolism of anandamide has previously been shown to underlay the biological actions of anandamide in bovine and sheep coronary arteries and rabbit platelets (Pratt et al., 1998; Braud et al., 2000; Grainger and Boachie-Ansah, 2001).
Vanilloid receptor heterogeneity is one factor that could explain why anandamide, olvanil, and resiniferatoxin are weaker agonists in main bronchi than in mesenteric arteries. Radioligand binding studies indicate that multiple capsaicin-sensitive vanilloid receptors may indeed exist, because capsazepine was 35- to 50-fold more potent as an inhibitor of specific [3H]resiniferatoxin binding in the airways than in spinal cord, dorsal root ganglion, and urinary bladder (Szallasi et al., 1993). As shown in the present study, Schild plots for capsazepine using capsaicin and resiniferatoxin as agonists yielded pA2 values that did not differ significantly between main bronchi and mesenteric arteries. Unless capsazepine is unable to discriminate between these subtypes of receptors, these findings suggest that main bronchi and mesenteric arteries are endowed with a single population of vanilloid receptors. The slope coefficients of the Schild plots were in all cases greater than unity, which may indicate that not only the agonists but also capsazepine binds to its receptor in a cooperative manner (Szallasi et al., 1999).
A carrier-mediated uptake of anandamide by a hypothetical anandamide membrane transporter (AMT) is considered to be essential for termination of the biological effects of anandamide (Di Marzo et al., 1994; Beltramo et al., 1997). Di Marzo and coworkers recently reported that the effect of anandamide on vanilloid receptors in HEK293 cells expressing TRPV1 is dependent on such an uptake mechanism (De Petrocellis et al., 2001a), supporting the idea that anandamide and capsaicin reach the binding site on the vanilloid receptor from the inside of the cell (Jung et al., 1999; Jordt and Julius, 2002). The present study provides pharmacological evidence that the AMT is present on primary afferents in blood vessels, but not in airways, and underscores the importance of this transport system for the biological action of fatty acid amides in native cells. Such variability in the expression of the AMT among different phenotypes of primary sensory nerve can help explain why anandamide and olvanil are more potent agonists in mesenteric arteries than in main bronchi. In line with this, the anandamide transport inhibitor AM404, which is a very potent activator of vanilloid receptors (De Petrocellis et al., 2000; Zygmunt et al., 2000), is also a poor constrictor of guinea pig airways (Craib et al., 2001; Tucker et al., 2001), but it is powerful as a vasodilator of guinea pig mesenteric arteries (D. A. Andersson, E. D. Högestätt, and P. M. Zygmunt, unpublished data). VDM13 did not affect responses to capsaicin in either tissue. This eliminates an effect of VDM13 on the vanilloid receptor (De Petrocellis et al., 2001a) or on subsequent steps of the signal pathways. It is also in line with capsaicin having a low affinity for the AMT (Melck et al., 1999). Our findings also raise the possibility that the biological activity of endogenous and synthetic fatty acid amides may be altered in conditions associated with up- or down-regulation of the AMT. Acidosis and activation of protein kinases A and C can increase the effect of anandamide on vanilloid receptors, which may be of relevance in the setting of inflammation (Premkumar and Ahern, 2000; De Petrocellis et al., 2001b; Olah et al., 2001; Vellani et al., 2001). Whether the AMT is regulated in a similar manner remains to be elucidated. In this context, it is interesting that nitric oxide, which may act as a cotransmitter in primary sensory neurons (Lundberg, 1996), enhances the cellular uptake of anandamide (Maccarrone et al., 1998;Maccarrone et al., 2000; De Petrocellis et al., 2001a).
Although VDM13 substantially reduced the differences in potencies of the agonists between the tissues, anandamide, olvanil and resiniferatoxin were still more potent in mesenteric arteries than in main bronchi. It is unlikely that the remaining potency differences reflect an incomplete inhibition of the transporter, because the concentration of VDM13 used in the present study (50 μM) completely inhibits the accumulation of anandamide in HEK293 cells (De Petrocellis et al., 2001a). The intrinsic efficacy and the receptor content are other factors determining the potency of an agonist. According to the classic receptor theory, the potency of an agonist will decrease with a decreasing number of receptors. However, an agonist with a low intrinsic efficacy may still induce a full response if the amount of receptors is large enough. Because there are some indications in the literature that capsaicin has a larger intrinsic efficacy than olvanil and resiniferatoxin at native vanilloid receptors (Wardle et al., 1997), we performed computer simulations to explore the possibility that agonists having different intrinsic efficacies are affected to various extents by changes in the receptor content. Allowing for variation of these parameters, the computer simulations could successfully predict the intriguing experimental observation that although capsaicin was equally potent in the two tissues, the other agonists were less effective in main bronchi than in mesenteric arteries. These findings are also in line with the conclusion that the agonists act on the same type of receptor in both tissues.
The present study shows that vanilloid receptor agonists may display varying degrees of tissue selectivity depending on the presence of a facilitated membrane transport mechanism and the magnitude of the receptor reserve. Accordingly, vanilloid receptor agonists, being good substrates for the AMT and having low intrinsic efficacy, will display selectivity toward tissues and bioassay systems expressing this transporter and having a large receptor reserve. Our findings help explain why airway obstruction is not uniformly observed after systemic administration of vanilloid receptor agonists and provide a conceptual framework for developing tissue-selective agonists devoid of bronchoconstrictor activity.
Footnotes
- Received March 11, 2002.
- Accepted June 3, 2002.
-
The Swedish Research Council, the Swedish Council for Planning and Coordination of Research, Swedish Society for Medical Research, and the Medical Faculty of Lund (ALF) supported this work. The Swedish Research Council supported P.M.Z.
Abbreviations
- AMT
- anandamide membrane transporter
- CB1
- cannabinoid receptor subtype 1
- FAAH
- fatty acid amidohydrolase
- NK1
- neurokinin subtype 1
- NK2
- neurokinin subtype 2
- PMSF
- phenylmethylsulfonyl fluoride
- VDM13
- arachidonoyl-5-methoxytryptamine
- TRPV1
- vanilloid receptor subtype 1
- HEK
- human embryonic kidney
- WIN 55.212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone
- HU-210
- Δ8-tetrahydrocannabinol dimethylheptyl
- SR 140,333
- (1-{2-[3-(3,4-dichlorophenyl)-1-(3-isopropoxyphenylacetyl)piperidin-3-yl]ethyl}-4-phenyl-1-azonia-bicyclo-[2.2.2.]octane chloride
- SR 141716A
- N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride
- SR 48,968
- (S)-N-methyl-N-[4-(4- acetylamino-4-phenyl piperidino)-2-(3,4-dichlorophenyl)butyl]benzamide
- U46619
- 9,11-dideoxy-9α,11α-methanoepoxy prostaglandin F2α
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}