Article Text

Abstract
Hereditary pancreatitis (HP) is a rare inherited disorder, characterised by recurrent episodes of pancreatitis often beginning in early childhood. The mode of inheritance suggests an autosomal dominant trait with incomplete penetrance. The gene, or at least one of the genes, responsible for hereditary pancreatitis has been mapped to the long arm of chromosome 7 and a missense mutation, an arginine to histidine substitution at residue 117 in the trypsinogen cationic gene (try4) has been shown to segregate with the HP phenotype. The aim of this work was to investigate the molecular basis of hereditary pancreatitis. This study was performed on 14 HP families. The five exons of the trypsinogen cationic gene were studied using a specific gene amplification assay combined with denaturing gradient gel electrophoresis (DGGE). The present paper describes three novel mutations, namely K23R and N29I and a deletion –28delTCC in the promoter region. We also found a polymorphism in exon 4, D162D. In eight of these families we found a mutation which segregates with the disease. A segregation analysis using microsatellite markers carried out on the other families suggests genetic heterogeneity in at least one of them. Our findings confirm the implication of the cationic trypsinogen gene in HP and highlight allelic diversity associated with this phenotype. We also show that the pattern of inheritance of HP is probably complex and that other genes may be involved in this genetic disease.
- hereditary pancreatitis
- mutation analysis
- cationic trypsinogen
Statistics from Altmetric.com
Hereditary pancreatitis is a rare autosomal dominant disorder characterised by recurring episodes of severe abdominal pain and is often experienced by young patients.1 The disease was first recognised by Comfort and Steinberg2 in 1952 and, since then, more than 100 kindreds have been published. Most of these families were white, although two have been reported as originating from Japan.3 The disease is the most common cause of recurrent pancreatitis from childhood and the clinical picture of the disorder includes recurrent episodes of pancreatitis, equal sex distribution, family history of at least two other affected members, frequent presence of calcified stones in the pancreatic ducts, and absence of known precipitating factors.1 Often patients develop complicated chronic pancreatitis, characterised by pseudocysts, chronic abdominal pain, diabetes mellitus, etc.
By using a positional cloning approach, we previously mapped the gene responsible for the disorder on the long arm of chromosome 7 at 7q354 and this localisation was independently confirmed by Whitcomb et al.5 Some potential candidate genes are located in the region, and among them eight trypsinogen genes are situated in the T cell receptor β chain gene (TCRβ) complex. Whitcomb et al 5 have since examined the cationic trypsinogen and found a missense mutation, an Arg to His change (R117H) that segregated perfectly with the phenotype in their large pedigree. This confirmed that the cationic trypsinogen gene (try4) is at least one of the genes responsible for hereditary pancreatitis.
In order to gain further insight into the molecular basis of this disorder, 14 families from different regions of France with members suffering from HP were studied. We looked for the presence of the R117H mutation in these families; when the R117H mutation was not found we scanned the coding sequence and the intron/exon junctions and part of the promoter region of the try4 gene by carrying out a denaturing gradient gel electrophoresis assay (DGGE). We observed three novel mutations that segregate with the disease. We also performed a segregation analysis with highly polymorphic microsatellite markers and discovered a family which exhibited possible genetic heterogeneity. These data not only show the allelic heterogeneity of hereditary pancreatitis, but also allow for new insights into the molecular basis of this disorder.
Materials and methods
FAMILY STUDIES
Each family member was examined by a physician to confirm their affected or unaffected status, with participants providing their informed consent. The diagnostic criteria were those proposed by Gross and Jones.6 According to the classical clinical definition, the patient experiences severe epigastric pain radiating to the back during an acute episode of pancreatitis, which is alleviated when in the fetal position. Symptoms in these families begin early in childhood, under 5 years of age. Calcification of the pancreatic gland can be shown by plain films of the abdomen and ultrasonography which shows gland enlargement. Computed tomography is often used for better delineation of certain features highlighted by ultrasonography.
DENATURING GRADIENT GEL ELECTROPHORESIS ANALYSIS
EDTA blood samples were collected and DNA prepared according to standard procedures. We have developed a PCR-SSP-DGGE approach. As the nucleotide sequences of the five trypsin genes are about 91% homologous, we compared the nucleotide sequences of the five genes and of the three pseudogenes in order to develop sequence specific primers. Specific amplifications of the try4 gene were obtained either by using two specific primers or by a combination of a group of specific primers (table 1). Two pairs of primers were designed to analyse exon 3.
Primers used
PCR amplification was performed for 35 cycles at 94°C for one minute, 68°C for one minute, and 72°C for one minute. For each exon, a GC clamp was attached to the 5′ end of one of these specific primers in order to analyse these specific amplification products using denaturing gradient gel electrophoresis. The amplification reaction for the DGGE analyses has been described elsewhere.7 8 Amplification products were run on 6.5% polyacrylamide gel containing a linear denaturing gradient from 50 to 100%. Electrophoresis was performed at 75 V for five to 11 hours depending on the melting map of the amplified sequence. PCR fragments displaying altered behaviour in the gel were then sequenced.
DNA SEQUENCING
Purified PCR products were sequenced directly by either the Cy5 autocycle sequencing kit (Pharmacia Biotech) or the thermo Sequenase sequencing kit (Amersham International) according to the manufacturer’s instructions. Sequences were analysed on an automated fluorescence based ALF Express sequencing system (Pharmacia).
MICROSATELLITE MARKERS
Amplification of each dinucleotide repeat (D7S684, D7S2505, D7S2468, D7S2513, D7S661, D7S676, D7S2511) was performed using a Perkin Elmer 9600 Thermocycler in a final volume of 50 μl containing 40 ng of genomic DNA, 50 pmol of each primer, 1.25 mmol/l dNTP, and 0.4 UTaq polymerase. The markers used for haplotype construction are presented in fig1.
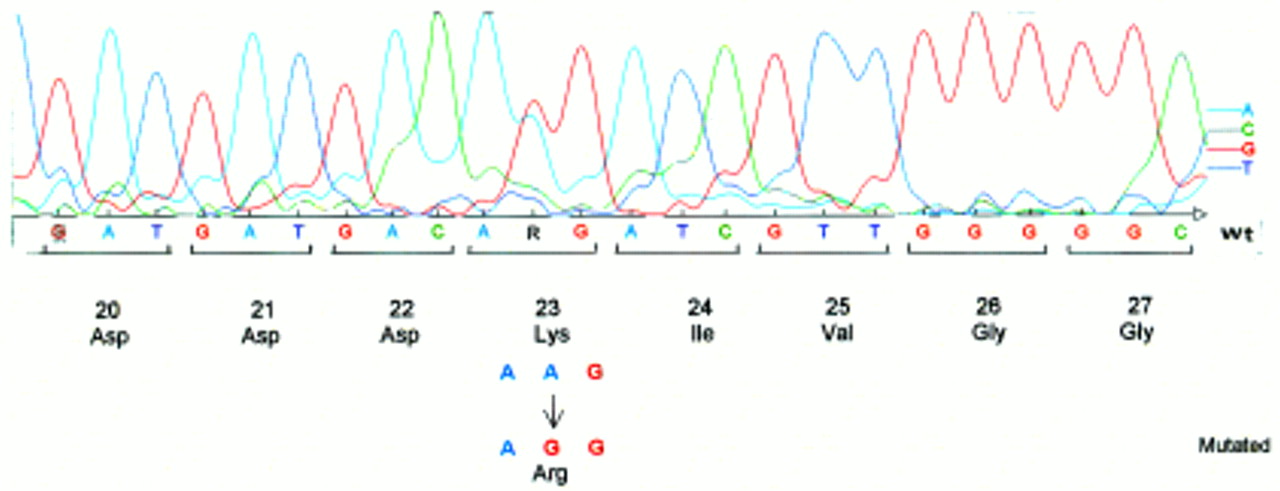
Illustration of the A→G transition at codon 23 which results in a lysine to arginine substitution at amino acid 23 (K23R).
Results
Fourteen families from different regions of France were studied. All contained at least three affected subjects; however, in four of these families only two subjects were studied. We first looked for the presence of the R117H missense mutation in the try4 gene. The discovery of this mutation allowed Whitcomb et al 5 to propose that the try4 gene is responsible for the disease. The R117H missense mutation was in fact found in our large initial kindred, an exceptional kindred4 of 249 members, which has been detailed elsewhere and which allowed us to map the HP gene to the long arm of chromosome 7. The R117H mutation was present in the 47 affected members of this family. This mutation has also been found in three other families in the present study. Segregation between the mutation and the phenotype was perfect in these four families. In the other 10 families in the study, the try4 gene was scanned in order to determine the molecular basis of hereditary pancreatitis in these cases.
The try4 gene is composed of five exons and is part of a 625 kb region of the genome whose sequence has been totally determined.9The region is highly duplicated as complete cloning of the TCRb locus has shown the presence of five trypsinogen genes and three pseudogenes. The five trypsinogen genes are highly homologous, displaying 95% homology and all residing in a 10 kb region duplicated in tandem. Moreover, the sequence homology in the coding region can be as high as 97%. We developed a denaturing gradient gel electrophoresis assay to scan each exon and its intron/exon boundaries. For this purpose, we designed a couple of primers specifically to amplify each exon; at least one of these primers was allele specific (see Materials and methods). This specificity was confirmed by the sequence data obtained for each exon.
DGGE analysis of exon 2 exhibited an altered profile of migration in three families. The direct sequencing of the PCR product showed evidence of two different nucleotide changes. The first one is an A→G transition at codon 23 that results in an arginine (AGG) to lysine (AAG) substitution at amino acid residue 23 (K23R), using the recent recommendations for nomenclature.10 The second nucleotide change is an A→T transversion which is predicted to result in an isoleucine (Ile) (ATC) to asparagine (AAC) substitution, namely N29I. This last mutation was found to segregate with the disease in two different unrelated families (fig 2). To rule out the possibility that these two mutations could result from a neutral polymorphism, we looked for these nucleotide changes in a large series of 200 DNA samples free from known genetic disorders and used as controls. None of these samples carried either of these two mutations. These results support the assumption that these mutated alleles are HP specific. We have also found in one patient an altered profile of migration in the PCR fragment encompassing the promoter region. Sequencing of the product showed a deletion of three bases, TCC at position –28 (from ATG). As we did not find any mutation in the gene coding sequence of six families studied (that is, 42% of the family studied here) we performed a segregation analysis with microsatellite markers (D7S640, D7S495, D7S684, D7S661, D7S676, D7S688) in order to study the transmission of haplotype linked to the HP gene. Family N illustrates the result of this segregation analysis: the two affected sibs (II.2 and II.3) have inherited two different haplotypes. However, III.2 did not receive her grandfather’s haplotype, indicating that, at least in this family, another gene located elsewhere in the genome could be responsible for the disorder (fig3).
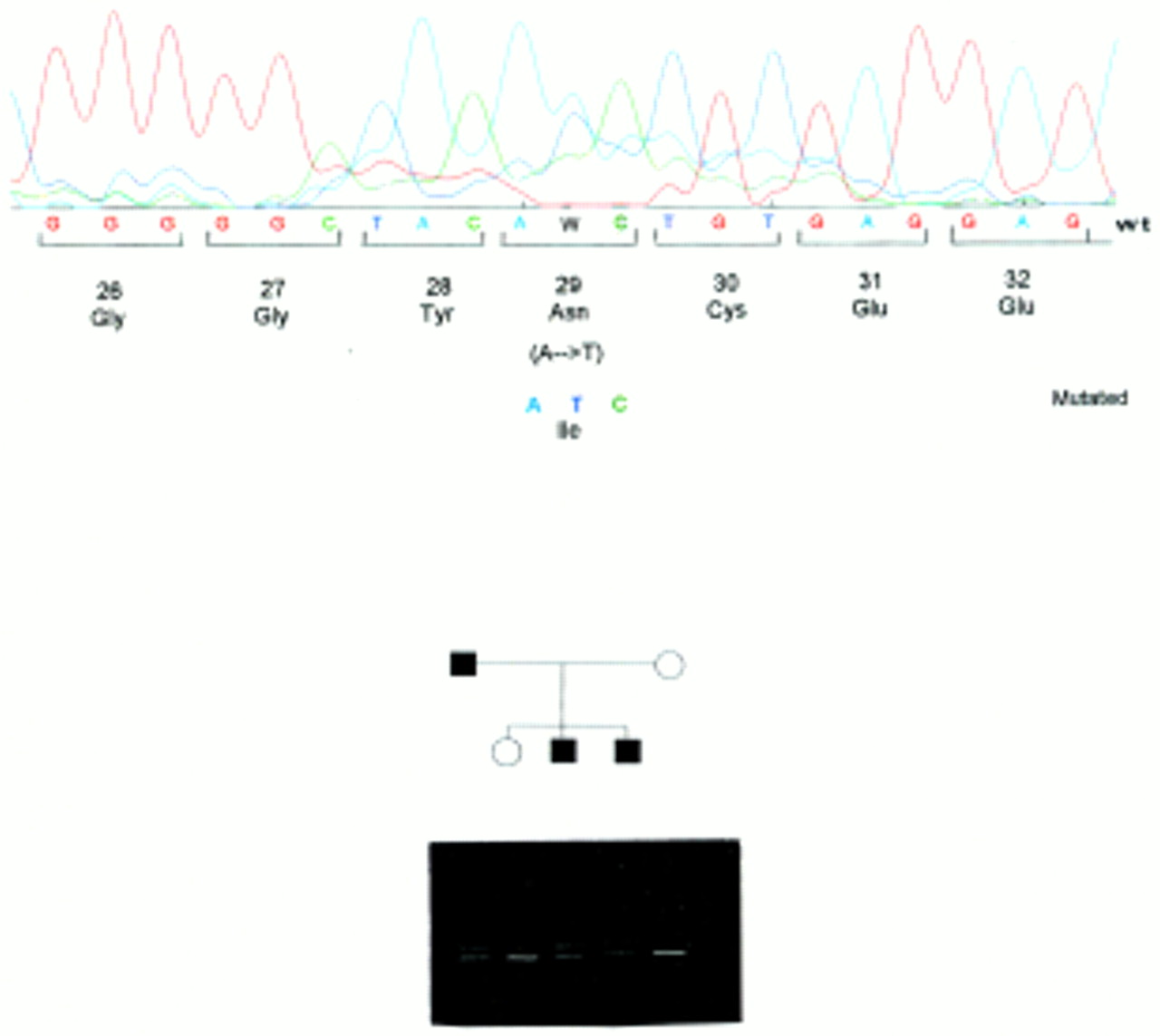
The A→T change leading to an isoleucine (ATC) to asparagine substitution (N29I). This mutation segregates with the disease in two families and the segregation of the DGGE pattern of migration is shown.

{kind=link}
{kind=link}
{kind=link}
Family N. Affected and unaffected subjects from this family are indicated by filled and open symbols, respectively. Genotypes of seven loci linked to the HP locus are shown in their map order. II.2 and II.3 were both affected with different maternal and paternal haplotypes, while III.2 did not inherit her grandfather’s haplotype.
Discussion
The results reported in this paper provide further evidence implicating the cationic trypsinogen gene in hereditary pancreatitis. Besides the R117H mutation, a missense mutation N21I was recently reported by Gorry et al.11 This mutation was found in one family and it was reported that the onset of symptoms was delayed and admissions to hospital were fewer in affected patients. In this study we observed three other mutations, K23R and N29I in exon 2 and −28delTCC in the promotor region of the try4 gene. The strategy used here to screen the gene (DGGE) is probably the most powerful technique for locating nucleotide variation within a gene. The capability of this DGGE technique to identify almost 100% of mutations in a gene has previously been reported; it enabled us to identify nearly 100% of mutations in the CFTR gene in certain populations.12 In the present study dealing with gene analysis in 14 HP families (table 2), we identified a mutation in only 42% of cases. Because the techniques used in this study are extremely sensitive, it may be hypothesised that, in the HP families with unidentified mutations, these mutations are outside the coding sequence of the gene, for example, in the introns or in the promoter region of the gene. It should be noted that in the eight families where we identified a mutation (four pedigrees with the R117H, one with the K23R, and two with the N29I, one with –28delTCC), the penetrance of the disease is complete. When the data from these eight families were pooled, the 72 affected patients over 29 years carried one mutation in the HP gene, whereas none of the healthy subjects older then 25 years carried a mutated allele. This observation indicates complete penetrance of the disease in families with an identified mutation. More than 100 families linked with HP have been reported since 1952. HP is considered an autosomal dominant disorder with variable expression and an estimated penetrance of 80%. Our results now suggest that inherited pancreatitis results from mutations in the try4 gene in more than 50% of cases, but that in some HP families try4 appears not to be involved. This suggests that other gene(s) or factor(s) located elsewhere in the genome are responsible for the low penetrance of HP.
Mutations in the 14 HP families
It is evident now that genetic counselling could be offered to these families. The four known mutations could be screened quickly and the patients informed of whether they were at risk of developing the disease. An appropriate prevention strategy could be proposed to any symptom free carrier of a mutated allele in the HP gene in order to reduce the occurrence of the condition.
Cationic trypsinogen is the precursor of trypsin, one of the principal enzymes secreted by the pancreas, with trypsin catalysing its own conversion from zymogen. When Whitcomb et al 5 identified the R117H mutation in HP affected patients, they suggested that the R117H mutation renders this site resistant to trypsin and trypsin-like proteases. It could thus prevent trypsin inactivation, consequently modifying enzyme activation/inhibition balance and resulting in pancreatic autodigestion. It is evident that the two mutations found in exon 2 and described in the present work act differently from R117H and it can be hypothesised that they alter the tertiary structure of the protein and the trypsin inhibitor binding site. Because the pancreatic secretory trypsin inhibitor (PSTI) normally inhibits up to 20% of trypsin activity, one would expect that if this regulatory mechanism is disrupted, excessive activity of trypsin would result in autodigestion and pancreatitis.13 14 This hypothesis can be confirmed by analysing these mutated forms of the cationic trypsinogen protein at the biochemical level.
The effect of the –28delTCC is only speculative at present. It could be expected that this deletion might modify and increase the level of try4 transcription and that a higher level of trypsin could be difficult to control by the different inhibitors, such as PSTI or other proteins resulting in pancreatic autodigestion.
Information derived from identification of the molecular basis of HP, along with an understanding of pathogenicity, should help us to progress towards a rational treatment for this disorder, not only for the rare hereditary forms of the disease, but perhaps for most cases of pancreatitis, including the sporadic or alcoholic forms.
Acknowledgments
This work was supported by the Programme Hospitalier de Recherche Clinique, the Direction Générale de la Recherche et de la Technologie - Jeune Equipe 2064, and by INSERM (CRI 96-07). We thank Arianne Le Menn for secretarial assistance.