Article Text

Abstract
Background: Hereditary mixed polyposis syndrome (HMPS) is characterised by colonic polyps of mixed histological types that are autosomal dominantly inherited and eventually lead to colorectal cancer (CRC). Study of the molecular basis of HMPS will enhance our knowledge of the genetic basis of the mixed polyposis-carcinoma sequence in both hereditary and sporadic CRC.
Methods/Results: We performed a genomewide linkage search on 15 members of a three-generation HMPS family using the GeneChip Human Mapping 10K Array and identified a 7 cM putative linkage interval on chromosome 10q23. Subsequently, 32 members from two HMPS families were typed with nine microsatellite markers spanning the region and the linkage was confirmed with a maximum multi-point logarithm of the odds (LOD) score of 4.6 (p<0.001). The 10q23.1–10q23.31 haplotypes segregate with the disease in both families. We screened for mutations in four candidate genes within the linkage region and identified an 11 bp deletion in the bone morphogenesis protein receptor 1A (BMPR1A) gene in one family.
Conclusions: Our results indicate that BMPR1A mutation accounts for HMPS. The data suggest that inactivating BMPR1A can initiate colorectal tumourigenesis via the mixed polyposis-carcinoma sequence.
- CRC, colorectal cancer
- FAP, familial adenomatous polyposis
- GDAS, Genechip DNA Analysis
- HMPS, hereditary mixed polyposis syndrome
- ISC, intestinal stem cell
- JPS, juvenile polyposis syndrome
- LOD, logarithm of the odds
- PCR, polymerase chain reaction
- SGH, Singapore General Hospital
- colorectal cancer
- haplotype
- linkage
- mixed polyposis
- whole-genome SNP genotyping
Statistics from Altmetric.com
- CRC, colorectal cancer
- FAP, familial adenomatous polyposis
- GDAS, Genechip DNA Analysis
- HMPS, hereditary mixed polyposis syndrome
- ISC, intestinal stem cell
- JPS, juvenile polyposis syndrome
- LOD, logarithm of the odds
- PCR, polymerase chain reaction
- SGH, Singapore General Hospital
It is generally accepted that colorectal carcinogenesis is a multi-step process and most colorectal cancer (CRC) is initiated by the inactivation of adenomatous polyposis coli (APC) or activation of β-catenin via the adenoma-carcinoma sequence.1 Recent evidence has also begun to unravel the genetic basis of the hamartoma-carcinoma sequence in rare syndromes with CRC manifestation such as Peutz-Jeghers, Juvenile, and Cowden’s.2 However, the initiating event for the mixed hyperplastic/serrated adenomatous polyposis-carcinoma sequence for colorectal carcinogenesis3–5 remains unclear.
Hereditary mixed polyposis syndrome (HMPS; MIM 601228) is characterised by colonic polyps of mixed hyperplastic, adenomatous, and occasional juvenile types and eventually leads to CRC. It is therefore an ideal model for dissecting the underlying genetic basis of the mixed polyposis-carcinoma sequence for both hereditary and sporadic CRC. HMPS was first described in a large Ashkenazi family, SM96.6 The syndrome is similar to familial adenomatous polyposis (FAP) in that it is autosomal dominantly inherited and the polyps can progress to CRC. Unlike the carpeting adenomas of FAP, however, the polyps in HMPS are of mixed histology, fewer in number, and appear to be confined to the large bowel. Furthermore, the syndrome is unlinked to APC, the gene responsible for FAP.
The HMPS locus was originally mapped to chromosome 6q16–21.7 Recently, however, this was shown to be incorrect.8 Using up-dated data from family SM96, and two additional Ashkenazi families, SM13119 and SM2592, the putative HMPS locus was remapped to a haplotype between markers D15S1007 and D15S118 on chromosome 15q13.8
Two Singapore Chinese families were diagnosed as having HMPS with similar pathological features as SM96. They also do not have APC germline mutations.10 Present study shows that the Ashkenazi haplotype is not associated with the disease locus in these two families. Whole genome screening with the GeneChip Human Mapping10 K Array followed by genomewide and localised linkage analyses with microsatellite markers were then performed.
METHODS
Sample collection and phenotypic classification
HMPS families 1 and 2 were registered with the Singapore Polyposis Registry in 1989. Peripheral lymphocyte specimens were collected from family members and spouses with informed consent over the past 10 years.
Clinicopathological data were retrieved from histology reports and patient medical records of Singapore General Hospital (SGH). Affection status was defined as the occurrence of three or more adenomatous, hyperplastic, or juvenile polyps or polyps of mixed histology of these three types. Individuals with no clinicopathological data were classified as unknown. Available histological sections (haematoxylin and eosin stained) were reviewed to characterise and classify the polyps.
This study was approved by the institutional review board of SGH.
Genomewide scan, haplotype construction, and linkage analysis
Genomic DNA was extracted using standard procedures. Whole-genome scan was performed with the Affymetrix GeneChip Human Mapping 10K Array Xba 131. A 250 ng sample of genomic DNA was digested with the restriction enzyme XbaI, amplified, fragmented, labelled, and hybridised to the 10K array as per the manufacturer’s instructions (Affymetrix, Santa Clara, CA). Arrays were then scanned with the Affymetrix 3000 scanner and analysed with GeneChip DNA analysis (GDAS) to generate a genotype call for each of the SNP probes on the array.
The SNP genotypes were imported by GDAS port into Merlin software for initial non-parametric analyses. Subsequent parametric analyses were carried out with GeneHunter (version 1.2) and Varia (version 1.0; Silicon Genetics, Palo Alto, CA) software. HMPS was modelled as a dominant trait (q = 0.001), with penetrances AA = 0.90, Aa = 0.90, and aa = 0.001. A logarithm of the odds (LOD) score of 3.3 or greater is taken as significant evidence of linkage, an LOD score of 1.9 or greater but less than 3.3 as suggestive linkage, and an LOD score of −2 or less as significant evidence of non-linkage.11 Marker allelic frequencies were from the genotyping of founders in both families as the allelic types and frequencies of the local population were distinct from the Caucasian population reported in the GBD Human Genome Database.
Genotyping and linkage analysis by microsatellite markers
Polymerase chain reaction (PCR) amplification of microsatellite markers was performed using 100 ng of genomic DNA in 25 μl reactions. After amplifications, PCR products were run on an ABI automated DNA sequencer. Results were analysed using Genotyper (Applied Biosystems, Foster City, CA) software.
Two-point and multi-point LOD scores were calculated using the GeneHunter (version 1.2) or LINKAGE program12 assuming the same autosomal dominant inheritance.
Sequence analysis
All coding exons and flanking sequences of BMPR1A, MINPP1, PTEN, and PCDH21 were amplified from genomic DNAs and cDNAs. Primer pair sequences are available upon request. Gene sequences were downloaded from the Ensembl database. PCR products were sequenced using the ABI automated DNA sequencer and BigDye Terminator Cycle Sequencing kit according to the protocol of the manufacturer.
RESULTS
Phenotypic features
Affected members of HMPS families 1 and 2 have colonic phenotypes very similar to that of SM96. The polyps showed hyperplastic, adenomatous, or juvenile-type morphology (fig 1). Juvenile-type polyps were documented in four individuals only: individuals 2 and 30 of family 1 and individuals 3 and 4 of family 2 (table 1). The juvenile-type polyps are associated with hyperplastic changes and hence are atypical juvenile polyps. One third of the patients were documented to have polyps with mixed juvenile and hyperplastic or mixed hyperplastic and adenomatous components on different visits. The mean age of diagnosis for all affected individuals was 32.4 years. More than half of the patients manifested with polyps throughout the large bowel. Three individuals from family 1 and three individuals from family 2 eventually developed CRC.
Colonic manifestation of HMPS patients

Multiple polyps of mixed morphology in individual 2 of family 1 (A–C) and individual 4 of family 2 (D–F). Panels (A–B) show sections of a mixed hyperplastic-juvenile polyp featuring (A) dilated, non-dysplastic glands (×40, original magnification) and (B) hyperplastic glands (×100, original magnification); panel (C) shows a separate mixed adenomatous-hyperplastic polyp and hyperplastic glands with serrated lumina indicated with an arrow (×100, original magnification). Panels (D–E) show sections of a mixed hyperplastic-juvenile polyp featuring (D) a dilated, non-dysplastic juvenile type gland (arrow) (×100, original magnification) and (E) a hyperplastic gland (arrow) with a serrated lumen (×100, original magnification); panel (F) shows a separate tubular adenoma with mildly dysplastic surface tubular glands (arrow) (×100, original magnification).
Extracolonic manifestation was rare in both families. Individuals 5 and 21 (mother and son) of family 1 were documented to have one right retinal lesion each although this was not substantiated with medical records. The only extracolonic manifestation documented for family 2 was in individual 8 who developed papillary thyroid carcinoma at the age of 40, 15 years after he was diagnosed with colonic polyps and cancer. No upper gastrointestinal polyps were documented in any individual.
Clinical history documented on three generations of these families indicated autosomal dominant inheritance (fig 2).
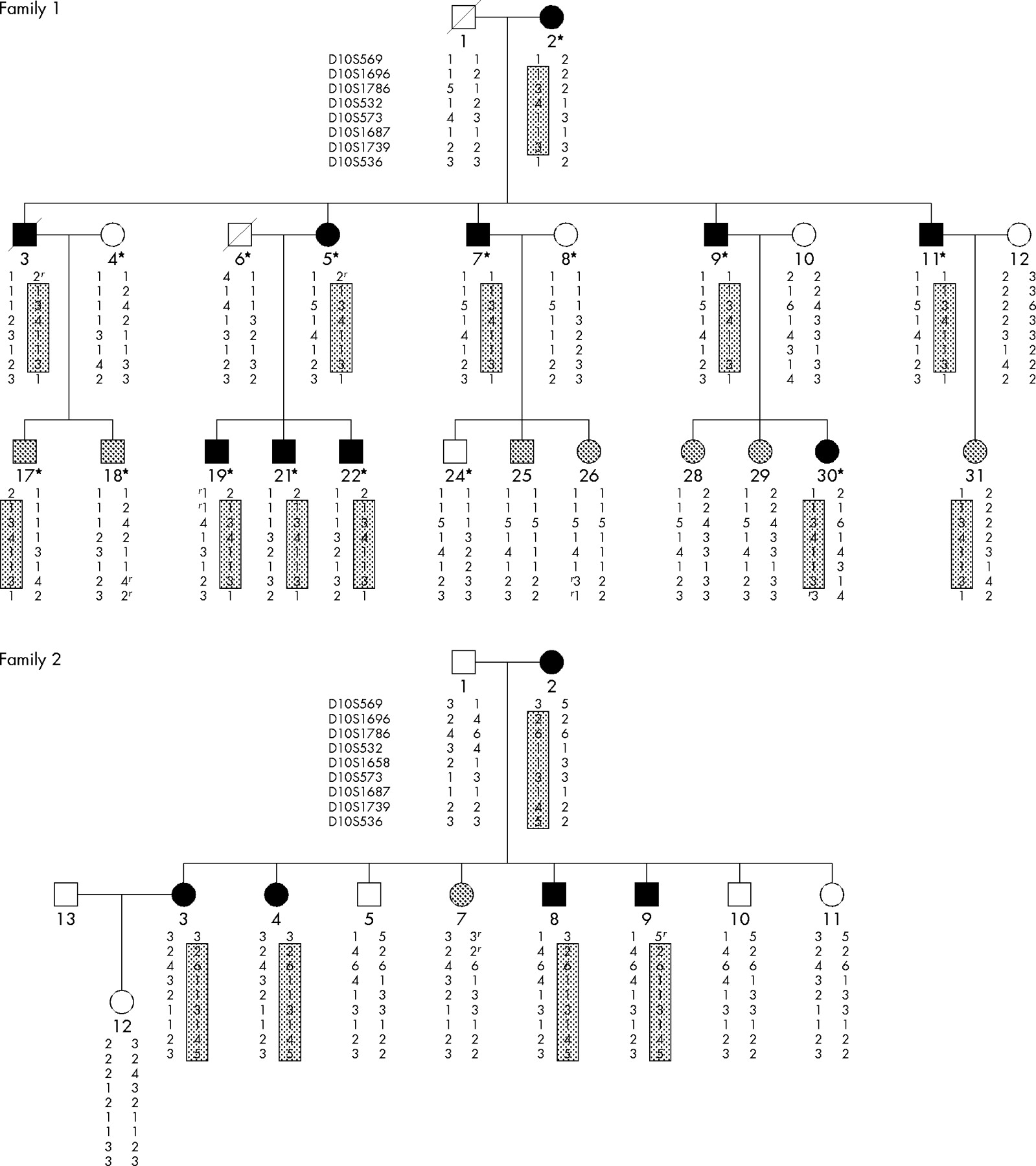
Pedigrees of families 1 and 2, showing haplotypes for chromosome 10q23 microsatellite markers. Asterisks denote individuals in family 1 who were genotyped by the high throughput SNP array. Closed, grey, and open symbols denote affected, unknown, and unaffected individuals, respectively. Boxed haplotype denotes the putative disease haplotype. The letter “r” besides the allele refers to recombination. Haplotypes for individuals 1 and 3 from family 1 and individual 1 from family 2 were inferred.
Exclusion of linkage to Ashkenazi haplotype
To investigate whether the Ashkenazi haplotype is responsible for the HMPS phenotype in the Singapore families, linkage analysis with microsatellite markers D15SS1010, D15S1007, ACTC, and D15S118 encompassing the putative HMPS locus in Ashkenazi families was performed on all 32 available individuals from both families. The maximum two-point and multi-point LOD scores at ACTC are 0.20 (at θ = 0.3) and −5.0, respectively, for the two Singapore families (data not shown).
Haplotype construction shows that the putative disease haplotype did not co-segregate with affected individual 11 of family 1 who was first diagnosed with tubular adenomas and on subsequent visits found to have mixed hyperplastic and tubular adenomatous polyps. For family 2, no definitive disease haplotype could be assigned.
Thus, linkage and haplotype analyses indicate that the HMPS locus for the two families is not located around marker ACTC as reported for the Ashkenazi families.
Genomewide linkage analysis
Whole genome scan with the GeneChip Human Mapping 10 K Array was then performed on 15 (nine affected, four unaffected, two unknown) individuals from family 1 (fig 2) to locate the HMPS locus. The mean signal detection and SNP call rates were 97.6% and 94.7%, respectively, indicating experimental integrity.
After filtering for non-Mendelian inheritance with the GDAS port, the rest of the SNP genotypes were used for linkage analysis. Initial analysis with Merlin revealed two very similar maximal non-parametric linkage scores of 12.9 (p<0.0001) and 12.7 (p<0.0001) in chromosomal regions 10q23 and 6q24, respectively. Subsequent analyses with GeneHunter indicated similar results. A maximal parametric multi-point LOD score of 2.4 was obtained at chromosome 10q23 (p = 0.002), defining a 7 cM region encompassing 27 SNPs. Another 3 cM region encompassing 16 SNPs on chromosome 6q24 achieved the identical LOD score of 2.4 (p = 0.002).
Further analysis was then performed with Varia using the haplotype based linkage analysis which provides more powerful statistics since haplotype blocks (which are more polymorphic) are used as markers rather than individual SNPs. Filtering for haplotype blocks with the highest LOD scores resulted in the same 7 Mb region on chromosome 10q23 delimited by the SNP markers rs265460 and rs536243 at 83.1 and 89.9 Mb, respectively (fig 3A). Two haplotype blocks in this region had LOD scores exceeding 2.6 (θ = 0). One other block had an LOD score of 2.3 (θ = 0). No other region with an LOD score greater than 1.9 was found in the genome. The region at 6q24 had a maximum LOD score of 1.8. We therefore excluded chromosome 6q24 from further study.
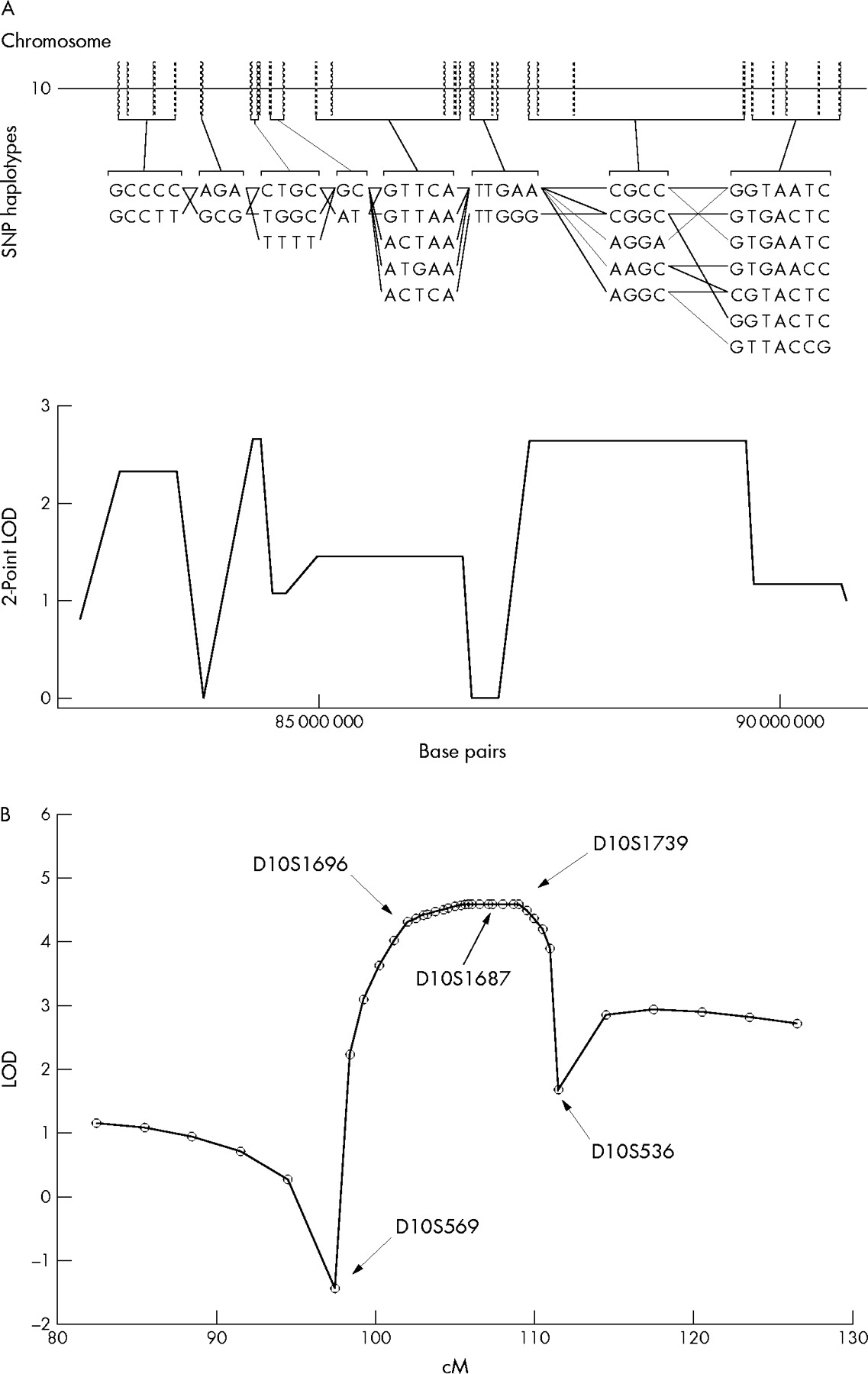
Genomewide (A) and chromosome 10q23 (B) linkage analyses. (A) SNP genotyping and haplotype based linkage analysis by Varia reveal a 7 Mb region on chromosome 10 (83–90 Mb) with LOD scores above 2.0. Each haplotype block contains a list of possible haplotypes as well as a pattern of co-transmission among the individuals (black lines) genotyped. The maximum haplotype based LOD score is 2.664 (θ = 0) at 84 Mb. (B) A sliding map of nine microsatellite markers was used to calculate multipoint linkage analysis between markers D10S569 and D10S536. Combined genotyping data from families 1 and 2 were used.
Microsatellite marker analysis at 10q23
Microsatellite markers encompassing chromosome 10q22–23 were then used to genotype all 32 individuals of both families. Nine microsatellite markers (D10S569, D10S1696, D10S1786, D10S532, D10S1658, D10S573, D10S1687, D10S1739, and D10S536) spanning the region from 97.4 to 111.5 cM were used. Pairwise linkage analysis between the disease locus and 10q markers was performed with the MLINK program of LINKAGE software. The maximum two-point LOD score obtained for both families combined was 3.03 (θ = 0) at D10S1739 (table 2). The maximum two-point LOD score for family 2 was 1.92 (θ = 0) at D10S536. However, recombination was observed in family 1 at this marker. Therefore, the HMPS locus is unlikely to be at this position. For both families, no recombination was observed between 102 and 109 cM indicating tight linkage between the HMPS locus and the region delimited by markers D10S1696 and D10S1739. Furthermore, the maximum multi-point LOD score is 4.6 (p<0.001) from D10S1696 to D10S1739 for the two families combined (fig 3B) suggesting significant evidence of linkage.
Two-point LOD scores for 10q22–23 markers
The putative disease haplotypes co-segregated with all affected individuals in both families (fig 2). It also segregated with individuals 17 and 31 of unknown status in family 1. Marker D10S569 (97.4 cM) had recombined out for affected individuals 3 and 5 of family 1 and individual 9 of family 2. Marker D10S536 (111.5 cM) had recombined out for affected individual 30 of family 1. Thus, haplotype construction also shows that the minimal region containing the HMPS gene lies between markers D10S1696 and D10S1739 from 102 to 109 cM or 83–90 Mb.
Germline mutation in BMPR1A causes HMPS
The critical interval of 83–90 Mb on 10q23 consisting of a panel of 19 characterised and 17 hypothetical genes (not shown) was scanned for promising candidates (fig 4A). Based on their reported roles in polyposis and tumour suppression, the bone morphogenetic protein receptor 1A BMPR1A,13–15 the tumour suppressor PTEN,16–18 multiple inositol polyphosphate phosphatase MINPP1,19 and the protocadherin 21 precursor PCDH2120 were selected for further analyses.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
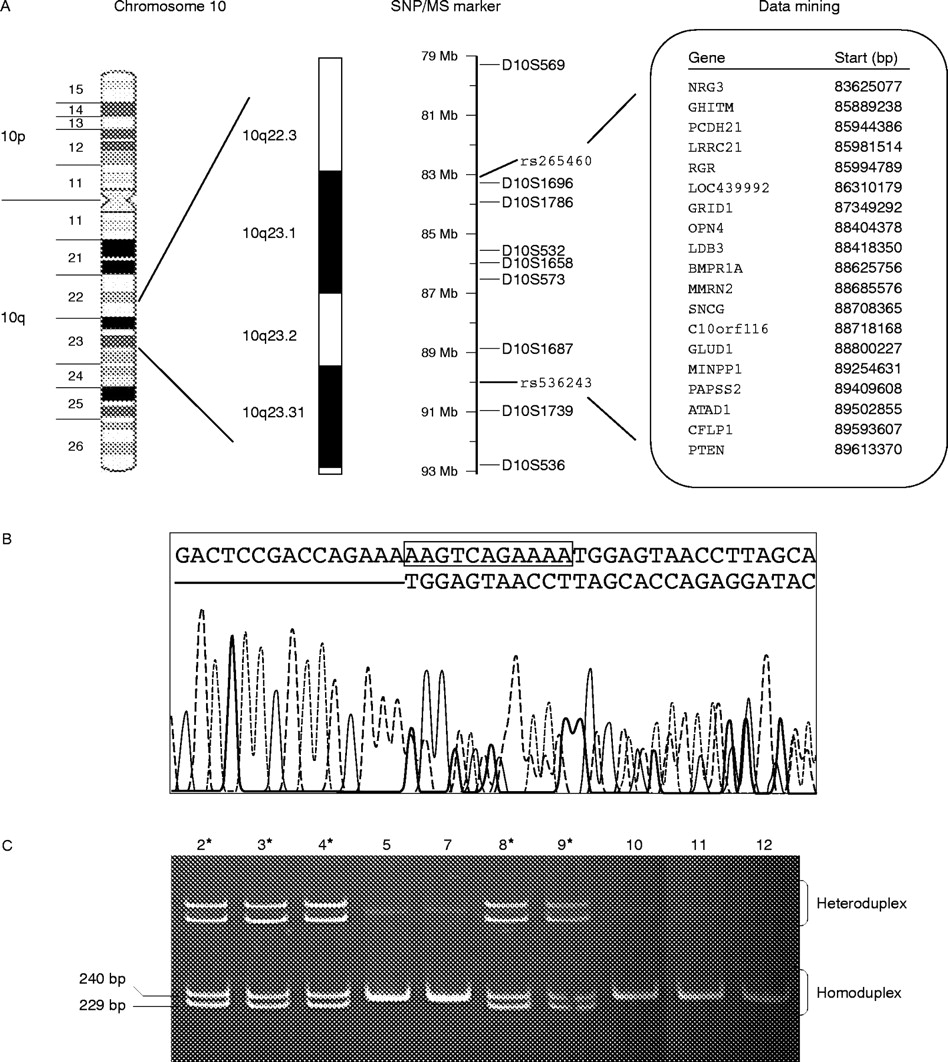
Mutation in BMPR1A causes HMPS. (A) The list of SNPs, microsatellite (MS) markers, and characterised genes in the critical interval of 83–90 Mb on 10q23. Genes in bold are genes with OMIM identification. (B) Sequence analysis showing BMPR1A 11 bp deletion at codon 43. (C) All affected individuals (asterisks) of family 2 show both homoduplex and heteroduplex bands of the amplified 240 and 229 bp fragments while all unaffected individuals exhibit the 240 bp homoduplex bands only when run on a 8% polyacrylamide gel.
Sequence analysis of all coding exons of BMPR1A performed on the genomic DNA of the proband (affected individual 2) of family 2 shows an 11 bp deletion at codon 42 of exon 2, resulting in a frameshift and truncation of translation (fig 4B). The truncation deleted all the functional domains of BMPR1A, including the Activin receptor domain in the 5′ extracellular region. The mutation is fully penetrant as further analysis with all available members of family 2 shows that all affected individuals harbour the mutation and no unaffected individuals inherited the mutation (fig 4C).
Direct sequencing analyses of all exons and flanking sequences of BMPR1A, PTEN, MINPP1, and PCDH21 on genomic DNAs and cDNAs of two affected individuals of family 1, however, reveal no detectable mutation in any of the four genes. Furthermore, the microsatellite markers flanking these candidate genes remain heterozygous for affected members whenever informative (fig 2 and fig 4A) indicating no large genomic deletions.
DISCUSSION
In this study, linkage analyses performed on two HMPS families using a genomewide SNP scan (fig 3A) followed by microsatellite markers analysis (table 2 and fig 3B) indicated significant linkage to a 7 Mb (83–90 Mb) region on 10q23 (fig 4A). The study illustrates the power of coupling whole genome SNP genotyping with haplotype based linkage analysis to locate a particular disease locus. A truncating mutation at the 5′ end of BMPR1A at 88.6 Mb was then shown to account for the HMPS phenotype in one family (fig 4B,C).
Germline mutation in BMPR1A was previously found to be associated with a subset of juvenile polyposis syndrome (JPS; MIM 174900) patients.13–15 The phenotypic features of the two families in this study, however, like SM96,6 differ from JPS. JPS usually affects infants or children between the ages of 5 and 15 years, manifesting with 50 or more juvenile polyps.16 In contrast, no classical or typical juvenile polyp was documented in any member of these two families (table 1). When present, atypical juvenile polyps were often seen together with hyperplastic components (fig 1 and table 1). Unlike JPS, the polyps were also not predominantly located in the left colon.
The detection of BMPR1A mutation in an HMPS pedigree raises the question whether HMPS and JPS could be (at least in part) allelic and represent different phenotypic expressions of mutation in the same gene. Just as germline mutations in APC can cause diverse phenotypic manifestations including those of distinct Turcot and Gardner syndromes, it is perhaps not surprising that mutation in BMPR1A could be responsible for two different syndromes which after all are based on pathological, not molecular, definitions.
No BMPR1A, PTEN, MINPP1, or PCDH21 germline mutation was detected in affected individuals of family 1. Other methods of inactivation of these four candidate genes which were undetected by present techniques (including possibly large intragenic deletion involving many exons) could be responsible for the HMPS phenotype in family 1. Alternatively, it is possible that another candidate HMPS gene could reside in the 10q23 region.
BMP signalling was recently shown to inhibit intestinal stem cell (ISC) renewal by suppression of the Wnt-β-catenin signalling pathway in a mouse model of JPS.21,22 The authors of that study showed that inactivation of the BMP pathway via the loss of function of its receptor, Bmpr1a, could lead to an increase in ISC number and crypt fission and hence clonal expansion of aberrant crypts (hyperplasia) through increased nuclear localisation of β-catenin in the ISCs. Deregulation in Wnt-β-catenin signalling is the initiating event in the adenoma-carcinoma pathway of colorectal carcinogenesis.1 In view of this recent evidence and the morphology of the HMPS polyps, it is conceivable that the loss of function mutation in the human homologue, BMPR1A, could initiate colorectal tumourigenesis via the mixed juvenile/hyperplastic/adenomatous polyposis-carcinoma pathway by functioning in an analogous manner.
Acknowledgments
The authors thank Ms Connie Ouyang, Dr Hui Peng, Dr Yi Zhao, and Ms Kim Yoong Puong for technical support.
REFERENCES
Footnotes
-
This work is funded in part by grants from the National Medical Research Council (NMRC/0638/2002) and the Biomedical Research Council (03/1/27/19/257) of Singapore
-
Competing interests: none declared