Article Text

Abstract
Background: In patients with juvenile polyposis syndrome (JPS) the frequency of large genomic deletions in the SMAD4 and BMPR1A genes was unknown.
Methods: Mutation and phenotype analysis was used in 80 unrelated patients of whom 65 met the clinical criteria for JPS (typical JPS) and 15 were suspected to have JPS.
Results: By direct sequencing of the two genes, point mutations were identified in 30 patients (46% of typical JPS). Using MLPA, large genomic deletions were found in 14% of all patients with typical JPS (six deletions in SMAD4 and three deletions in BMPR1A). Mutation analysis of the PTEN gene in the remaining 41 mutation negative cases uncovered a point mutation in two patients (5%). SMAD4 mutation carriers had a significantly higher frequency of gastric polyposis (73%) than did patients with BMPR1A mutations (8%) (p<0.001); all seven cases of gastric cancer occurred in families with SMAD4 mutations. SMAD4 mutation carriers with gastric polyps were significantly older at gastroscopy than those without (p<0.001). In 22% of the 23 unrelated SMAD4 mutation carriers, hereditary hemorrhagic telangiectasia (HHT) was also diagnosed clinically. The documented histologic findings encompassed a wide distribution of different polyp types, comparable with that described in hereditary mixed polyposis syndromes (HMPS).
Conclusions: Screening for large deletions raised the mutation detection rate to 60% in the 65 patients with typical JPS. A strong genotype-phenotype correlation for gastric polyposis, gastric cancer, and HHT was identified, which should have implications for counselling and surveillance. Histopathological results in hamartomatous polyposis syndromes must be critically interpreted.
Statistics from Altmetric.com
Juvenile polyposis syndrome (JPS, OMIM 174900) is an autosomal dominant disorder characterised by the occurrence of multiple juvenile polyps in the gastrointestinal tract, specifically in the stomach, small intestine, colon and rectum.1–3 Pathogenic germline mutations in the SMAD4 (MADH4) gene have been identified in around 20% of patients with JPS, and another 20% of patients were found to exhibit a mutation in the BMPR1A gene.1 4–6 A higher frequency of gastric polyposis in carriers of SMAD4 mutations compared with carriers of BMPR1A mutations has been reported.6–8 Most SMAD4 or BMPR1A germline mutations published to date are small insertions/deletions and single base substitutions leading to nonsense, splice-site or missense mutations (Human Gene Mutation Database). Recently, germline deletions encompassing the contiguous genes BMPR1A and PTEN on chromosome 10q have been reported in five cases of juvenile polyposis of infancy.9 10 These deletions have been found by absence of parental alleles in the children, quantitative PCR or fluorescence in situ analysis.
Large genomic deletions or duplications encompassing ⩾1 exons have been found in several genes using the multiplex ligation-dependent probe amplification (MLPA) assay.11–15 Using the recently developed MLPA test kit for quantitative evaluation of genes involved in JPS, we examined whether large SMAD4 or BMPR1A deletions or duplications are present in patients with JPS with as yet unknown germline mutations, and verified the genotype–phenotype correlation with respect to gastric polyposis.
PATIENTS AND METHODS
Subjects
In total, 80 unrelated patients were examined for mutations in the SMAD4, BMPR1A and PTEN genes (table 1). Of these, 65 patients met the clinical criteria for JPS (“typical JPS”) as described by Jass16: >5 colorectal juvenile polyps, juvenile polyps throughout the gastrointestinal tract, or any number of juvenile polyps and a positive family history. Of the remaining 15 patients, designated as “presumed JPS”, 8 had only a presumptive diagnosis of JPS on the basis of isolated JPS polyps (1–3 juvenile polyps in the absence of a family history of JPS); in 3 patients, a florid gastric juvenile polyposis in the absence of colorectal juvenile polyps was described; and in 4 patients, clinical information was incomplete. The diagnosis of JPS was based on pathology reports; in 25 of the 80 families, the histological results of the polyps were re-reviewed by an experienced gastrointestinal pathologist. Of the 65 index patients meeting the clinical criteria of JPS, 25 (39%) came from families where⩾2 generations had been affected. In five cases, a de novo mutation was found, but in another 11 patients, no further familial cases of JPS were known. No information on the family history was available for 24 patients. Clinical characteristics, molecular genetics and family history of the patients are provided in detail in table 2. Of the 80 patients, 30 have been included in previous reports.6 17–19
Detection of point mutations
Genomic DNA was extracted from peripheral EDTA-anticoagulated blood samples according to standard salting-out procedure. Analysis for small mutations in the SMAD4, BMPR1A, PTEN and CDH1 genes was performed by direct sequencing of the entire coding regions of the genes on an ABI Prism 377 or ABI 3100 automated sequencer (Applied Biosystems, Darmstadt, Germany) using the BigDye terminator V.2.0 or V.1.1 cycle sequencing kit.6 All germline mutations were confirmed in two independent PCR products. The numbering of the cDNA bases was carried out according to the reference sequences given in GenBank NM_005359.3 (SMAD4), NM_004329.2 (BMPR1A), NM_000314.4 (PTEN) and NM_004360.2 (CDH1), respectively, where +1 corresponds to the A of the ATG translation initiation codon.
SMAD4 transcript analysis
Fresh venous blood samples (2.5 ml) were collected into PAXgene Blood RNA tubes (PreAnalytiX; Qiagen, Hilden, Germany) containing RNA stabilising solution. Total RNA was extracted by use of the PAXgene Blood RNA Kit (Qiagen) according to the manufacturer’s protocol. First strand cDNA was synthesised from 2–3 μg of total RNA by random hexamer-primed reverse transcription with the Superscript First Strand System for RT-PCR (Invitrogen GmbH, Karlsruhe, Germany) according to the manufacturer’s protocol. Reverse transcriptase PCR (RT-PCR) fragments were obtained according to a standard PCR protocol, using a forward primer localised in exon 6 and a reverse primer in exon 10. RT-PCR products were separated on a 2% agarose gel and visualised with ethidium bromide on an ultraviolet imaging system (Biorad, San Diego, California, USA). Individual bands were excised from the gel and eluted with the High Pure PCR Product Purification Kit (Roche, Basel, Switzerland). Eluted DNA was reamplified with the same pair of primers and sequenced as described above.
Detection of large genomic deletions by MLPA
Large deletions or duplications were searched for with the MLPA assay for JPS. The newly developed MLPA Test Kit (SALSA P158-JPS; MRC-Holland, Amsterdam, The Netherlands) contains 15 paired probes from the SMAD4 region (1 probe for the promoter region, 3 probes for the first two noncoding exons and 11 probes for the coding exons), and 17 probes from the BMPR1A region (3 probes for the first 2 noncoding exons and 14 probes for 10 of the 11 coding exons). No probe could be designed for coding exon 5 of the BMPR1A gene because of its high homology to the BMPR1A pseudogene. The MLPA kit also contains nine probes for the coding region of the PTEN gene. Deletion screening was performed according to the manufacturer’s protocol. Briefly, 75 ng of genomic DNA in 5 μl TE buffer was heat-denaturated for 5 min at 98°C and hybridised overnight (16 h) at 60°C with the set of MLPA probes. Next, hybridised products were ligated (at 54°C) and amplified by PCR (30 cycles), and PCR fragments were separated on an ABI 3100 capillary sequencer.
Data were analysed using GeneMapper V.4.0 software (Applied Biosystems, Darmstadt, Germany) and gene dosage was calculated using the Coffalyser V4 program (MRC-Holland). All identified deletions were confirmed in a second independent reaction. Where possible, segregation of the deletions with the disease in the families was examined.
Statistical analysis and calculation of splicing efficiencies
The statistical comparison of features was performed with the Fisher exact test for categorical variables (presence of gastric polyposis) or the Student t test for continuous variables (age at diagnosis; age at gastroscopy). SPSS V.14.0 (SPSS, Chicago, Illinois, USA) was used for all statistical analyses. A p value of <0.05 (two-sided) was considered to be significant. Splicing efficiencies in the normal and mutant sequences were calculated using the splice prediction program of the Berkeley Drosophila Genome Project (BDGP).
RESULTS
Point mutations in SMAD4 and BMPR1A
Using direct sequencing of individual exons, we identified 17 germline mutations in SMAD4 and 13 mutations in BMPR1A in the 80 patients, resulting in an overall mutation detection rate of 38%, or of 46% when only the unequivocal clinical cases were included (table 1). To our knowledge, 12 of the mutations have not been described previously (table 2).
SMAD4 point mutations
Of the 17 SMAD4 point mutations, 11 were predicted to lead to truncated proteins and were thus considered definitely pathogenic (5 nonsense, 6 frameshift mutations). Moreover, four missense mutations were localised at highly conserved amino acid positions and were thus considered likely to be disease-causing (table 2). Two of the four missense mutations (patients JUV-14 and JUV-78) were proven to have occurred de novo. In a third patient (JUV-81), only a faint mutant signal (c.1082G→A;p.Arg361His) was found during sequencing of exon 8, suggesting that this mutation was present as a mosaic. The same sequencing pattern was obtained in a second blood sample from the patient and was confirmed in a PCR product generated with primers localised outside the first primer pair used in the regular diagnostic setting. Thus, the faint signal was not due to unequal allele amplification based on a variant in the primer sequence. Moreover, sequencing of a DNA sample isolated from a polyp confirmed the presence of the mutation at a slightly higher level (data not shown). Both parents of this patient were reported to have no polyposis.
The mutation c.1139G→A in exon 8 of the SMAD4 gene (JUV-44) is predicted to result in a missense mutation (p.Arg380Lys). However, this substitution, localised to the last position of the exon, interfered with splicing: a loss of the normal splice site (decrease in the splicing efficiency from 0.45 to <0.01) was predicted by the BDGP splice prediction program. Using mRNA analysis, we could show that the substitution led to the formation of a cryptic splice site localised within exon 8, resulting in a deletion of nucleotides 1003–1139 and formation of a premature stop codon due to a frameshift (fig 1). Thus, the correct designation of the mutation is c.1139G→A;r.1003_1139del137;p.Gly336AlafsX11. The PCR product obtained on mRNA with a forward primer localised within the deletion contained only the wild-type nucleotide (G at position 1139), showing that no full-length mRNA fragment was obtained from the mutant allele (not shown).

The variant c.425–6A→G in intron 2 of the SMAD4 gene (patient JUV-51) was predicted to create a new splice acceptor site and might thus be pathogenic. Unfortunately, no mRNA was available from this patient.
BMPR1A point mutations
Of the 13 point mutations identified in BMPR1A, five were nonsense, 2 frameshift, 4 missense and 2 splice site mutations (table 2). One of the splice site mutations (JUV-48) encompassed a deletion of 65 nucleotides localised to intron 4 of BMPR1A and included the highly conserved position −2 of the splice acceptor site of exon 5 (c.432-2_432-66del). This mutation was observed in two affected patients (mother and child). The variant was found because exons 4 and 5 were examined in the same PCR fragment. To date, no mRNA has become available for examination of the real effect on splicing.
LARGE DELETIONS IN SMAD4 AND BMPR1A
All patients without identified point mutation (50) and patients with missense mutations or as yet unspecified variants (10) were examined by MLPA for the presence of large deletions or duplications.
LARGE SMAD4 DELETIONS
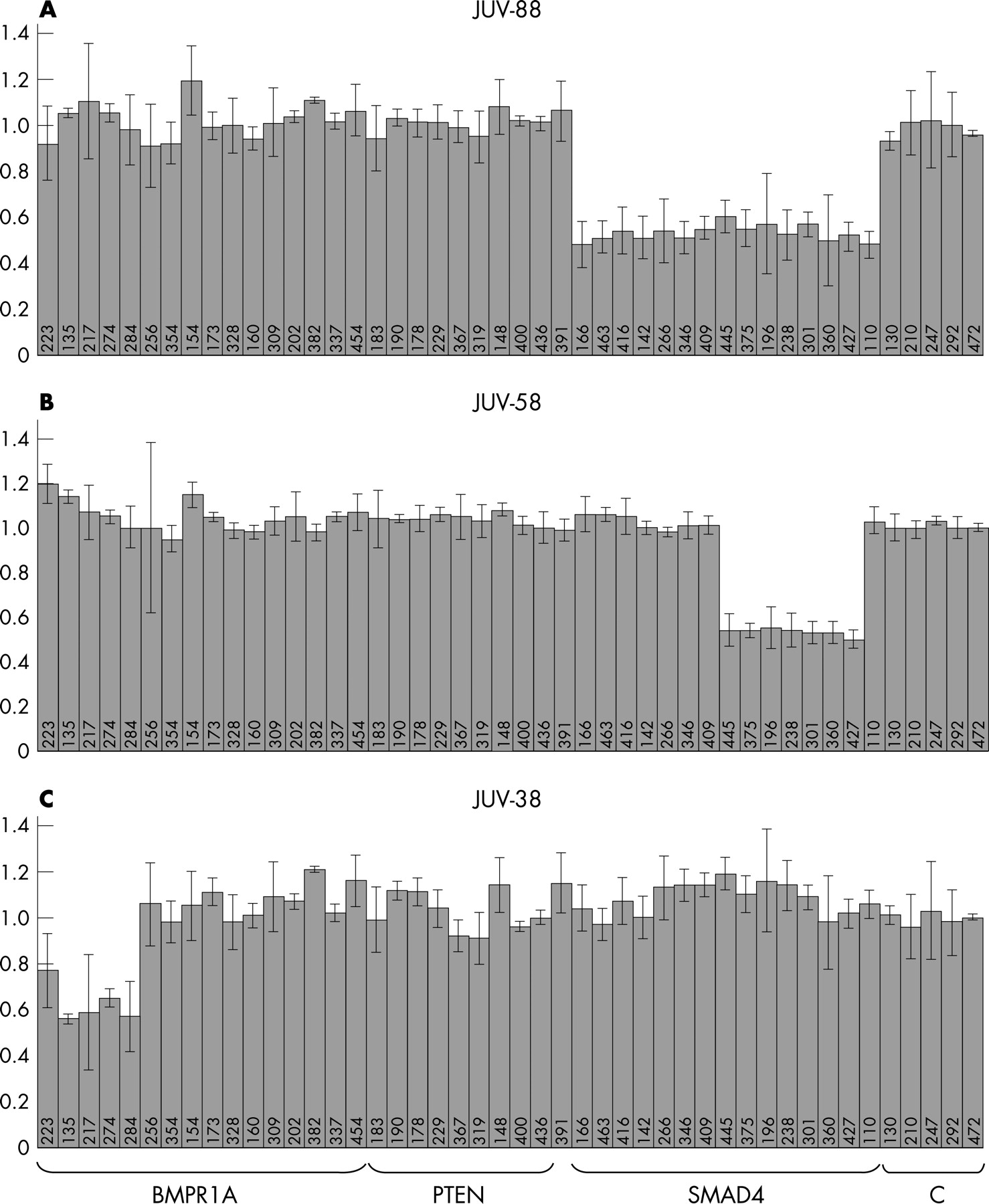
Large SMAD4 deletions were found in six patients. Four exhibited a heterozygous deletion of all SMAD4 probes encompassing the entire SMAD4 gene and the promoter region. One patient had a deletion of coding exons 5–11 and another had a deletion of coding exons 6–11 (fig 2). All deletions were confirmed in a second independent MLPA test. In one of the families (JUV-54), the deletion of the entire SMAD4 gene found in the index patient was confirmed in three other affected family members. The MLPA test kit readily found the large SMAD4 deletions in the 6 patients, whereas the remaining 54 patients and 5 normal controls revealed reproducible normal SMAD4 patterns with calculated relative values between 0.8 and 1.2.

{kind=link}
{kind=link}
LARGE BMPR1A DELETIONS
Deletions in the BMPR1A gene were found in three patients. One patient (JUV-38) had a deletion of four BMPR1A probes (the two first noncoding exons and the two probes designed for the first coding exon of the gene (table 1, fig 2). A heterozygous deletion of the two probes for coding exon 1 was found in patient JUV-22 and his affected father. Owing to the large introns localised to both sides of exon 1 (37 kb and 14.2 kb, respectively), we were unable to verify this deletion by long-range PCR on genomic DNA. In one patient (JUV-26), a deletion of the entire BMPR1A and PTEN genes was observed. The clinical phenotype of this patient and details of the deletion will be reported elsewhere.
Given the high homology between the BMPR1A gene and a pseudogene, reliability for BMPR1A is not as good as for SMAD4. In particular, wide variability was found with the MLPA probes designed for BMPR1A coding exon 4 and exon 10 (MLPA fragments of 154 and 382 bp, respectively), often showing nonreproducible relative peak heights of between 0.5 and 3.
MUTATIONS IN PTEN
In patient JUV-16, the MLPA test revealed an isolated “deletion” of PTEN exon 7. Sequencing this exon, we found the heterozygous nonsense mutation c.697C→T;p.Arg233X localised close to the hybridisation site of the MLPA probe for PTEN exon 7. The mutation was also found in the affected father of the index patient.
Subsequently, all the remaining mutation-negative patients were screened for PTEN germline mutations by direct sequencing. Of the 40 patients, 1 (JUV-18) was found to have a pathogenic splice site mutation in intron 4 (c.253+1G→T).
GENOTYPE–PHENOTYPE CORRELATION
The colorectal phenotype of SMAD4 and BMPR1A mutation carriers was indistinguishable: There was no significant difference in the median age at diagnosis of JPS between carriers of the SMAD4 (12 years) and the BMPR1A (14 years) mutations (p = 0.48; table 3). Both groups had a comparable number and histological spectrum of colorectal polyps.
Gastric polyposis
In a previous study on 29 unrelated patients with JPS with 12 identified mutations, we found an over-representation of gastric polyposis among carriers of SMAD4 mutations compared with carriers of BMPR1A mutations.6 A similar trend was observed when only the 27 patients (22 families) with known status of gastric polyposis who had not been analysed in our previous study were considered: 11 of 17 patients with SMAD4 mutations, but none of 11 patients with BMPR1A mutations, had gastric polyposis (p<0.01). In the combined sample (previously and newly analysed cases) information on results of gastroscopy was available for 30 patients with SMAD4 mutations (20 unrelated index patients and 10 affected relatives) and for 13 patients with BMPR1A mutations (nine index patients and four affected relatives) (table 3). Of the 30 patients with a SMAD4 mutation, 22 (73%) were found to have gastric polyposis. In contrast, only 1 of the 13 patients (8%) with BMPR1A mutations had gastric polyps (p<0.001). The over-representation of gastric polyposis in SMAD4 mutation carriers remained true even when age at gastroscopy was considered. Although the median age at gastroscopy was 35 (range 11–60) years for patients with SMAD4 mutations and 26 (range 4–73) years for patients with BMPR1A mutations, the difference was not significant (p = 0.71) (table 3).
Generally, gastric polyposis in SMAD4 mutation carriers is diagnosed later in life (median age at diagnosis 41 years) compared with diagnosis of colorectal polyps (12 years) (p<0.001). The difference in age at gastroscopy between SMAD4 mutation carriers with and without gastric polyps was highly significant: gastric polyps were diagnosed at a median age of 41 years, whereas patients without gastric polyps had gastroscopy at a median age of 16 years (p<0.001). No significant difference in the age at gastroscopy was observed between SMAD4 and BMPR1A mutation carriers without gastric polyposis (p = 0.15).
Gastric cancer
Consistent with this over-representation of gastric polyposis, all seven cases of gastric cancer were reported in families with SMAD4 mutations (index patient JUV-55; one affected relative in families JUV-55 and JUV-37; four affected relatives in family JUV-4). The brother of index patient JUV-4 had an early tubular adenocarcinoma diagnosed at 38 years of age; the histology results of the other three affected family members (two uncles and an aunt) were not available. In the relative of JUV-37, an early gastric cancer of the diffuse–infiltrating type surrounded by hyperplastic tissue was found at 42 years of age. This woman died from an adenocarcinoma of the small bowel. Index patient JUV-55 had an adenocarcinoma, and his brother had a well-differentiated adenocarcinoma diagnosed within a juvenile polyp.
To exclude a germline E-cadherin mutation as underlying cause of gastric cancer, mutation analysis of the CDH1 gene was performed in the affected brother of index patient JUV-4. No mutation was identified. DNA was not available from the other six patients.
Hereditary haemorrhagic telangiectasia
In addition to gastrointestinal polyposis, 5 of the 39 index patients with identified germline mutations (JUV 14, 44, 51, 58, 78) had a clinical diagnosis of hereditary haemorrhagic telangiectasia (HHT, Osler–Weber–Rendu disease). All five patients belong to the 23 index patients harbouring a SMAD4 mutation, thus the frequency of HHT among SMAD4 mutation carriers is 22% (5/23) in our sample.
Large deletions versus point mutations
No significant difference with respect in age at diagnosis between carriers of point mutations and large deletions of each gene (SMAD4: p = 0.80; BMPR1A: p = 0.12) was found; however, the statistical analysis in BMPR1A mutation carriers was limited because of the small number of patients with BMPR1A deletions (n = 3). In addition, the difference in presence of gastric polyposis (p = 0.3) and HHT (p = 1.0) between carriers of SMAD4 deletions and point mutations was not significant.
POLYP HISTOLOGY AND DIFFERENTIAL DIAGNOSES
The documented histological results of removed colorectal polyps varied considerably among patients as well as between different examinations in the same patient (table 2). Adenomatous components including dysplasia (intraepithelial neoplasia according to the revised World Health Organization classification) were described in many juvenile polyps. In addition, in the majority of patients with proven germline mutations in the SMAD4 or BMPR1A genes, presence of juvenile polyps (with or without intraepithelial neoplasia), hyperplastic polyps, pseudopolyps and adenomas were reported to different extents. In many cases, the initial histological results delayed the diagnosis of JPS; in some cases, juvenile polyps were only diagnosed when the tissue blocks were re-evaluated by an experienced pathologist. Similar diagnostic difficulties were evident for gastric polyps.
The accompanying infiltrate often leads to the assumption of inflammatory pseudopolyps, thus ulcerative colitis was a common initial diagnosis in our patients with JPS. Rare differential diagnoses include Morbus Ménétrier (giant hypertrophic gastritis) (patient JUV-36) and Cronkhite–Canada syndrome (CCS) (patient JUV-88). The latter patient, with a deletion of the entire SMAD4 gene, had been diagnosed at age of 12 years due to numerous polyps throughout the entire colon (diagnosed histologically as inflammatory pseudopolyps, granulation tissue polyps or juvenile polyps), severe anaemia and protein-losing enteropathy. Gastroduodenoscopy showed normal findings.
In both patients harbouring a germline PTEN mutation (JUV-16, JUV-18) a variety of different polyp types was reported, encompassing juvenile, hyperplastic, adenomatous and inflammatory polyps, although JPS was diagnosed in JUV-16 after histological re-evaluation by an experienced pathologist (table 2). Patient JUV-18 presented with additional extraintestinal tumours; he had a renal cell carcinoma and an intramuscular mixed benign tumour in the gluteal region composed of a lipoma and a haemangioma component.
DISCUSSION
Proportion of large deletions in JPS
In a comprehensive mutation screen of 80 unrelated patients with JPS, we identified point mutations in the SMAD4 and BMPR1A gene in 38% of patients (30/80 families) which is consistent with previous findings,6 7 20 or in 46% of patients when only the 65 typical cases were considered.
Before this study, the frequency of large genomic deletions in patients with JPS was unknown. Using the recently developed MLPA test kit we identified large SMAD4 and BMPR1A deletions in 26% (9/35) of the remaining mutation-negative patients who fulfilled the clinical diagnostic criteria of JPS and in 14% (9/65) of all patients with typical JPS, respectively. Neither point mutations nor large deletions were found in any of the 15 presumed JPS cases. The identification of large deletions in SMAD4 and BMPR1A genes increases the mutation detection rate to 49% (39/80) in all patients or to 60% (39/65) when only patients meeting the clinical JPS criteria are considered (table 1).
Overall, the MLPA test kit SALSA P158-JPS was proven to be an easily performed and reliable method to identify large genomic deletions in the SMAD4, BMPR1A and PTEN genes, although analysis of exon 4 and 10 of the BMPR1A gene was limited because of nonreproducible results.
Genotype–phenotype correlation
In a previous study, we observed a higher frequency of gastric polyposis cases among carriers of SMAD4 mutations compared with BMPR1A mutation carriers.6 We were able to confirm this higher frequency in a second independent sample of patients with JPS (p<0.01). In the entire sample, the frequency was 73% among SMAD4 mutation carriers. The occurrence of gastric cancer in this group of families alone reflects the same trend. The lifetime incidence of gastric polyposis in SMAD4 mutation carriers is probably higher, as the latest gastroscopy in mutation carriers without gastric polyps was performed at a significantly lower age than in mutation carriers with gastric polyps.
An underestimation of gastric polyposis in BMPR1A mutation carriers cannot be completely excluded, as age at gastroscopy did not differ significantly between the BMPR1A and SMAD4 mutation carriers without gastric polyps. However, it was clear that no SMAD4 mutation carrier without gastric polyps was older than 33 years, whereas all BMPR1A mutation carriers who underwent gastroscopy at advanced age (42, 48, 52 and 73 years) had no gastric polyps. The only BMPR1A mutation carrier with positive gastroscopic finding had only two juvenile polyps at 31 years of age. Moreover, there was no family history of gastric polyposis or gastric cancer in any of the BMPR1A mutation carriers.
Although HHT, an autosomal dominant disorder of vascular dysplasia, is usually caused by germline mutations in the ENG or ACVRL1 (ALK1) genes,21 there are several case reports of patients with combined symptoms of HHT and JPS.19 22–25 Recently, mutations in ENG were described in two patients with juvenile polyposis.10 Gallione et al identified SMAD4 mutations in all 14 examined patients from 7 unrelated families with the combined phenotype (JPHT; OMIM 175050).26 Our findings confirm this result and suggest that the phenotypic overlap of JPS and HHT is more common than previously thought (around a quarter of our index patients with JPS and SMAD4 mutations). Accordingly, JPS patients with SMAD4 mutations should be screened for typical HHT features, particularly vascular lesions, to avoid serious complications such as aortic aneurysm and pulmonary thrombosis. Similarly, patients diagnosed with HHT should be screened for gastrointestinal polyposis to ensure appropriate management.
Histopathological results and differential diagnosis
The diagnosis of JPS is based mainly on the presence of juvenile polyps in the gastrointestinal tract. Thus, histopathological evaluation is essential for correct classification and analysis of the appropriate genes. However, the histological findings documented in the medical records of the 80 index patients and their affected relatives encompassed a wide distribution of different polyp types in both SMAD4 and BMPR1A mutation carriers (“mixed polyposis”). Thus, polyp heterogeneity comparable with that described in patients with hereditary mixed polyposis syndromes (HMPS) seems to be a common feature in patients with JPS and reflects both the real occurrence of different polyp types and an uncertainty in histological assessment. Accordingly, in some patients, various diagnoses were initially reported, including ulcerative colitis, hyperplastic polyposis, unclear mixed polyposis or CCS.
Although juvenile polyps have a distinct morphology, small tumours and incomplete biopsies in particular may be a challenge. The presence of inflammatory infiltrates and intraepithelial neoplasia is a consistent feature of juvenile polyps. In all of our 13 cases with identified pathogenic germline mutation in which initially only hyperplastic, adenomatous, or inflammatory polyps had been diagnosed, juvenile polyps were uncovered by an experienced pathologist, who had been asked for a second opinion (table 2). This was particularly true for the gastric polyposis, even though appropriate diagnostic criteria have been defined. Our observation and that of others demonstrates the need for a critical interpretation of histopathological results in patients with gastrointestinal polyposis.10 19
Owing to the striking histological overlap between JPS and HMPS, the existence of a clinically defined HMPS is questionable. As a fraction of our mutation-positive patients with JPS would also have fulfilled the poorly defined criteria for HMPS, a substantial number of HMPS cases seem actually to be histopathological variants of JPS. Our data support the conclusion of Cao et al. that JPS and HMPS might be (at least in part) allelic entities,27 suggesting that particularly HMPS2 is neither clinically nor genetically a distinct condition.
One patient was referred for mutation analysis with the diagnosis of CCS, a rare and poorly delineated entity with a significant phenotypic overlap to JPS. It is usually described as late-onset, sporadic, non-inherited gastrointestinal polyposis of unknown aetiology accompanied by diffuse skin pigmentation, alopecia and onychodystrophy.28 29 As underlined by our patient, a proportion of polyposis cases might be misdiagnosed as sporadic CCS. In particular, young age at diagnosis and absence of typical ectodermal manifestations in patient JUV-88 argues against CCS.30
PTEN mutations
Mutation analysis of the PTEN gene in the 41 SMAD4 and BMPR1A mutation-negative patients revealed a germline mutation in two (5%); one of the mutations was found by chance using MLPA. Both patients (JUV-16, JUV-18) had a variety of different polyp types; one case presented with extraintestinal tumours not typical for JPS. Our results are in accordance with those reported by Sweet et al, who identified two PTEN germline mutations in 23 patients (9%) with a mixture of hyperplastic and adenomatous polyps. In both cases the reviewed clinical phenotype revealed features reminiscent of Cowden syndrome.10
Genetic heterogeneity and phenotype overlap in hamartoma syndromes is well known.31–33 Some polyposis patients with germline PTEN mutations are reported to harbour only juvenile polyps without extraintestinal lesions;32 however, critical histological re-evaluation, complete physical examination and medical history often reveals evidence for the diagnosis of Cowden syndrome, in particular, as the penetrance of this syndrome is low in patients <20 years of age.31 34 Consequently, although some patients with germline PTEN mutations present with clinical signs not diagnostic for Cowden syndrome, they should be classified as having PTEN hamartoma tumour syndrome and medically managed as such.10 31
Electronic database information
JPS: Online Mendelian Inheritance in Man (OMIM) 174900
SMAD4: OMIM *600993; reference sequence GenBank NM_005359.3; GI34147555
BMPR1A: OMIM *601299; reference sequence GenBank NM_004329.2; GI41349436
PTEN: OMIM *601728; reference sequence GenBank NM_000314.4; GI110224474
CDH1: OMIM *192090; reference sequence GenBank NM_004360.2; GI14589887
Human Gene Mutation Database, Cardiff: http://www.hgmd.cf.ac.uk/ac/index.php
Berkeley Drosophila Genome Project (BDGP): http://www.fruitfly.org/seq_tools/splice.html
GeneReviews: http://www.genetests.org/
In summary, we show that large SMAD4 and BMPR1A deletions are present in about 14% of all patients meeting the clinical criteria for JPS. We confirm a strong genotype–phenotype correlation regarding gastric polyposis, gastric cancer and HHT in SMAD4 mutation carriers, which should have implications for counselling and clinical surveillance. In case of any uncertainty or doubtful histological findings, re-evaluation of the polyp tissue by an experienced pathologist is recommended. As a correct clinical and molecular diagnosis has important consequences for appropriate clinical management, our observations and those of others underline the need for a more extensive analysis of the SMAD4, BMPR1A and PTEN genes in patients with hamartomatous polyposis including mixed polyposis syndromes.
Acknowledgments
We are grateful to the patients who participated in the study. The study was supported by the German Cancer Aid (Deutsche Krebshilfe e.V. Bonn, project no. 106244).
REFERENCES
Footnotes
Competing interests: None declared.
- Abbreviations:
- BDGP
- Berkeley Drosophila Genome Project
- CCS
- Cronkhite–Canada syndrome
- HHT
- hereditary haemorrhagic telangiectasia
- HMPS
- hereditary mixed polyposis syndromes
- JPS
- juvenile polyposis syndrome
- MLPA
- multiplex ligation-dependent probe amplification
- OMIM
- Online Mendelian Inheritance in Man
- RT
- reverse transcriptase