Article Text

Abstract
BACKGROUND Cystic fibrosis (CF) is characterised by an excess of free proteinases that destroy lung tissue. Despite this, previous studies have shown that patients with CF with a mild deficiency variant of the proteinase inhibitor α1-antitrypsin have less, rather than more, severe pulmonary disease. Alpha1-antichymotrypsin is another important serine proteinase inhibitor that protects the lung against proteolytic attack, and point mutations in the α1-antichymotrypsin gene that result in plasma deficiency are associated with chronic obstructive pulmonary disease.
METHODS The effect of α1-antichymotrypsin deficiency and the –15 α1-antichymotrypsin signal peptide genotype on lung function was assessed in patients with CF.
RESULTS One hundred and fifty seven patients with CF were screened and 10 were identified with a plasma deficiency of α1-antichymotrypsin (plasma concentration <0.2 g/l). In a multivariate analysis these individuals had significantly less severe lung disease than those who had normal or raised levels of α1-antichymotrypsin: forced expiratory volume in one second (FEV1) 69.9% predicted versus 53.2% predicted (p=0.04) and chest radiographic score of 7.2 versus 9.7 (p=0.03) for those with and without α1-antichymotrypsin deficiency, respectively. The –15 signal peptide genotype did not affect plasma levels, but the –15 Ala/Ala signal peptide genotype was over-represented in individuals with CF compared with healthy blood donor controls.
CONCLUSION These data indicate that deficiency of α1-antichymotrypsin is associated with less severe pulmonary disease in patients with CF, and support our previous observations that mild genetic deficiency of a proteinase inhibitor is associated with an improved outcome.
- cystic fibrosis
- α1-antichymotrypsin
- α1-antitrypsin
Statistics from Altmetric.com
Cystic fibrosis (CF) is characterised by a progressive destructive bronchitis secondary to chronic infection and inflammation. Genetic modifiers lying outside the cystic fibrosis transmembrane regulator (CFTR) gene are thought to account for some of the wide variability in pulmonary disease seen in individuals with the same CF mutation.1 ,2 The neutrophil derived proteinases elastase and cathepsin G are found in excess in sputum from individuals with CF3 and play a pivotal role in the associated pulmonary damage.3 ,4 We have previously shown that patients with CF and mild to moderate deficiency phenotypes of the proteinase inhibitor α1-antitrypsin have preserved lung function compared with those with a normal α1-antitrypsin phenotype.5 Moreover, CF patients with α1-antitrypsin deficiency were not over-represented among those who had died in childhood or who had undergone lung transplantation.6 This suggests that mild deficiency of the proteinase inhibitor permits downregulation of the inflammatory response, thereby reducing the severity of the pulmonary damage.7
Alpha1-antichymotrypsin is a 68 kDa serine proteinase inhibitor secreted by the liver, monocytes, and astrocytes.8 ,9 It is an inhibitor of cathepsin G and mast cell chymase10 and has been implicated in the modulation of immunological and cellular functions.11 ,12 Hereditary deficiency of α1-antichymotrypsin has a gene frequency of 0.003 in the Swedish population13 and is associated with liver disease and a raised residual volume on lung function testing.14 ,15 It is now recognised that this deficiency results from the 55Leu→Pro and 229Pro→Ala point mutations which reduce plasma levels in heterozygotes to 59% of normal. Both of these mutations have been associated with chronic obstructive pulmonary disease (COPD).16 ,17
The –15 Thr and Ala α1-antichymotrypsin signal peptide genotypes are present in almost equal quantities in the normal population. The –15Ala/Ala genotype is thought to increase the secretion of α1-antichymotrypsin relative to the–15Thr allele.18 Baseline levels of α1-antichymotrypsin are not affected by the polymorphism16 but the effect of the bi-allelic polymorphism in the acute phase has yet to be established.
We report here the effect of α1-antichymotrypsin deficiency and the –15 α1-antichymotrypsin signal peptide genotype on pulmonary disease severity in patients with CF. Moreover, we have determined the effect of the –15 signal peptide genotype on plasma levels of α1-antichymotrypsin.
Methods
CLINICAL DETAILS
One hundred and fifty seven patients with CF were recruited from two regional CF centres in Cambridge and Manchester, UK as previously described.5 Details of pulmonary function, pancreatic status, smoking history, liver function, age at onset of colonisation with Pseudomonas aeruginosa andBurkholderia cepacia, and α1-antitrypsin phenotype were recorded from patient interview and from the case notes. Colonisation was defined as two consecutive positive sputum cultures six months apart. Patients were classified as “pancreatic sufficient” (PS) and “pancreatic insufficient” (PI) according to their need for pancreatic enzyme supplements, and liver disease was determined from clinical features, liver biochemistry, and liver imaging. The severity of pulmonary disease was gauged from the patient's best forced expiratory volume in one second (FEV1) in the previous six months and was compared with age/sex matched normal controls19 to give a percentage predicted value of FEV1. A chest radiograph taken when the patient was stable was assessed using the Northern scoring system20 by a radiologist and a CF pulmonary physician who were blind to the identity of the patient. The Northern scoring system scores a posteroanterior chest radiograph in quadrants out of a maximum of 20, with higher scores representing more marked radiographic abnormalities. Cystic fibrosis genotypes were determined by the Department of Clinical Genetics at Addenbrooke's Hospital, Cambridge and the Department of Molecular Genetics, The Royal Manchester Children's Hospital, Manchester.
SERUM α1-ANTICHYMOTRYPSIN AND α1-ANTITRYPSIN
Serum α1-antitrypsin and α1-antichymotrypsin concentrations were measured by immunoturbidimetry by the Department of Biochemistry, Hinchingbrooke Hospital, Huntingdon against standards of known concentration. The reference ranges for α1-antichymotrypsin and α1-antitrypsin concentrations were 0.2–0.6 g/l (within batch coefficient of variation 5.5%) and 0.7–1.7 g/l (within run coefficient of variation 2.84%), respectively.
PRIMER SEQUENCES FOR α1-ANTICHYMOTRYPSIN SEQUENCING
Dideoxy chain termination sequencing of the α1-antichymotrypsin coding sequence and the signal peptide sequence was performed by cycle sequencing with33P-labelled dideoxy nucleotides and Thermosequenase (Amersham Life Science, Cleveland, OH, USA). Polymerase chain reaction (PCR) of the individual exons was performed using 1 μl DNA, 1.5 mM Mg2+, and 12.5 pmol of each primer in a reaction volume of 25 μl. Products were generated after 40 cycles at 94°C for 30 seconds, 50°C for 30 seconds, and then 40°C for 40 seconds for all exons. The primers used were as described by Faber and colleagues.16
- Exon II: 5′-TCCATCTggCCCTCTgAgACT-3′, CTCTACAgTggTTTgCCTAATAAC-3′;
- exon III: 5′-TATgAggACTCTgggCACTTC-3′, 5′-CCTgTATgAgTTCTCCCACCA-3′;
- exon IV: 5′-gCAggTAggTACTgATCAgCA-3′, 5′-CCAAAAgTCCATCTggCCTTC-3′;
- exon V: 5′-TgCgCATCTgTgTTTCCCgTg-3′, 5′-TgTgAgAgCTTCACAgggAAT-3′.
The PCR product was then incubated withexonuclease I and shrimp alkaline phosphatase according to the manufacturers' instructions, and sequenced by 33P cycle sequencing using Thermosequenase. One μl of PCR product was used for cycle sequencing with denaturation at 94°C for one minute, annealing at 45°C for one minute, and extension at 60°C for six minutes. Sequencing reactions were run on 7 M 8% w/v PAGE.
DETERMINATION OF THE –15THR→ALA α1-ANTICHYMOTRYPSIN SIGNAL PEPTIDE GENOTYPE
This was performed by PCR followed byBstNI restriction enzyme digestion. PCR was performed using 1 μl DNA, 1.5 mM Mg2+, 12.5 pmol of each primer in a 25 μl volume with denaturation at 94°C for 20 seconds, annealing at 50°C for 20 seconds, and extension at 74°C for 30 seconds. The primers used were 5′-TCCATCTggCCCTCTgAgACT-3′ and 5′-ggTTgAACTTgAggCCTTTgA-3′. Digestion was then carried out overnight with BstN1 at 60°C using 8 μl of the PCR product, the buffers provided, BSA and dH2O in a total volume of 20 μl according to the manufacturer's instructions. The products were run on 3% w/v agarose gels and were visualised by staining with ethidium bromide. Digestion of the 158 bp PCR product resulted in two distinct 86 bp and 72 bp products for an Ala homozygote, 158 bp, 86 bp, and 72 bp products for an Ala/Thr heterozygote, and a 158 bp product for a Thr homozygote. The products were run on 3% w/v agarose gels and visualised by staining with ethidium bromide. Genotyping was confirmed by sequencing a random selection of 12 individuals.
STATISTICAL ANALYSIS
Cystic fibrosis provides an ideal condition in which to test the effect of the –15 α1-antichymotrypsin signal peptide genotype on serum α1-antichymotrypsin concentration as many patients with CF have an inflammatory response that may be affected by the –15Thr→Ala signal peptide genotype. Alpha1-antitrypsin and α1-antichymotrypsin are both acute phase proteins. Previous studies have shown a close relationship between α1-antitrypsin and α1-antichymotrypsin.21 ,22 Indeed, we have recently shown that there is a linear relationship between plasma concentrations of α1-antichymotrypsin and normal M α1-antitrypsin over a range of concentrations.5 The serum concentration of α1-antitrypsin was therefore used to assess the inflammatory response in each individual. The effect of the –15 α1-antichymotrypsin signal peptide genotype on serum α1-antichymotrypsin levels was assessed by analysis of covariance, adjusting for the α1-antitrypsin level and phenotype.
One way analysis of variance and Fisher's exact test for frequencies were used to test for differences in patient characteristics between the factor groups studied. In addition, Pearson's χ2test was used to compare the proportions of CF patients with the different –15Thr→Ala signal peptide genotypes, with and without α1-antichymotrypsin deficiency, and a group of healthy controls.
The effect of α1-antichymotrypsin deficiency and the –15 α1-antichymotrypsin signal peptide genotype on FEV1% and chest radiographic score formed the primary investigation for this study. It was not possible to assess the effect of these factors on age at onset of colonisation withP aeruginosa because of insufficient numbers. Analysis of covariance was used to assess the effect of the factors on the outcome measures while adjusting for covariates that could possibly influence the relationship between the factors and the outcome measures. For each factor, both the observed mean (unadjusted for covariates) and the adjusted mean were calculated with the relevant tests performed on the latter. The covariates considered were age, sex, pancreatic status, P aeruginosa colonisation status, B cepacia colonisation status, CF genotype, smoking status, liver cirrhosis, body mass index, recruitment centre, and α1-antitrypsin phenotype.
Results
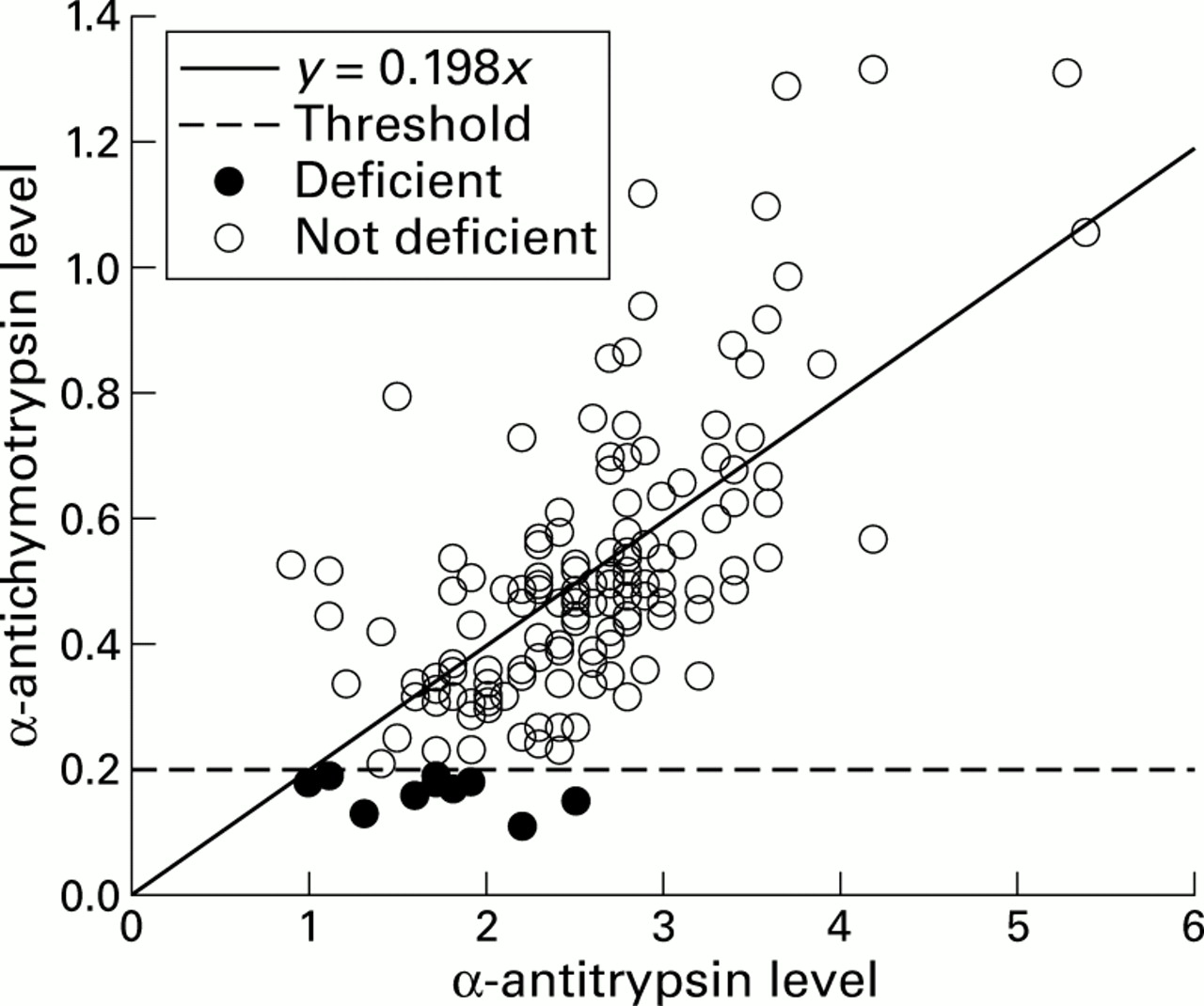
Ten patients a with plasma deficiency of α1-antichymotrypsin were identified from 157 subjects with CF (fig 1). All 10 had plasma α1-antichymotrypsin levels below the reference range (<0.2 mg/ml, mean 0.166 mg/ml). In addition, their α1-antichymotrypsin levels were inappropriately low for the degree of inflammation as assessed by comparison with their plasma α1-antitrypsin levels. Six of these 10 patients had plasma α1-antitrypsin levels at or above the normal reference range for α1-antitrypsin (1.7 mg/ml), indicating that they were mounting a vigorous inflammatory response. Two of the remaining four patients had α1-antitrypsin deficiency; one had the MS phenotype and the other was homozygous for the S allele. These patients would be expected to have baseline plasma levels of 80% and 60%, respectively, compared with the M homozygote, and would not be expected to have α1-antitrypsin levels above the normal range. DNA sequencing of the signal peptide and entire coding sequence of the α1-antichymotrypsin gene in these 10 cases did not reveal any point mutations when compared with the normal sequence.

Relationship between the plasma levels of α1-antichymotrypsin and α1-antitrypsin in patients with CF (r= 0.69, p<0.001). Ten patients with CF were identified with plasma α1-antichymotrypsin levels that were less than 0.2 g/l, despite the fact that six had a vigorous inflammatory response as indicated by α1-antitrypsin levels of more than 1.7 mg/ml.
The patient characteristics are shown in table 1. Interestingly, patients with α1-antichymotrypsin deficiency were less likely to be colonised with P aeruginosathan those with normal levels of α1-antichymotrypsin (30% versus 81%, p=0.001).
Characteristics of the study groups
EFFECT OF α1-ANTICHYMOTRYPSIN DEFICIENCY ON CF LUNG DISEASE
CF patients with α1-antichymotrypsin deficiency were found to have significantly less severe lung disease than those with normal levels of α1-antichymotrypsin (FEV169.9% predicted versus 53.2% predicted, p=0.04, table 2). Statistical analysis showed that this effect was independent of the age of the patient and colonisation with P aeruginosa. Those with α1-antichymotrypsin deficiency also had less severe radiographic changes (radiographic scores of 7.2 and 9.7 in those with and without α1-antichymotrypsin deficiency, respectively, p=0.03).
Effect of deficiency of α1-antichymotrypsin and the three different −15 α1-antichymotrypsin signal peptide genotypes: Ala/Ala, Thr/Thr and Ala/Thr on FEV1 % and the Northern chest radiography score
–15THR→ALA SIGNAL PEPTIDE GENOTYPE
The –15Thr allele (fig 2) was found in 47% of the control (healthy blood donor) population but in only 30% of those with α1-antichymotrypsin deficiency and CF (table 3). The incidence of the –15Thr allele did not differ between individuals with CF with and without α1-antichymotrypsin deficiency. However, there was a significant increase in the incidence of the –15Ala/Ala signal peptide genotype in patients with CF compared with controls (p=0.02). The –15 α1-antichymotrypsin signal peptide genotype had no effect on plasma levels of α1-antichymotrypsin (table 4). Interestingly, the –15Ala/Ala signal peptide genotype was associated with a better percentage predicted FEV1 and chest radiography score (p=0.004 for both; table 2).

{kind=link}
{kind=link}
3% w/v agarose gel showing the determination of the–15Thr→Ala α1-antichymotrypsin signal peptide genotype. Lane 1, DNA size markers; lanes 2, 4, 5, 7,–15Ala homozygote; lanes 3, 6, 9, –15Ala/Thr heterozygote; lane 8, –15Thr homozygote.
Frequencies of the−15Thr→Ala signal peptide genotypes in healthy controls and in CF patients with and without plasma α1-antichymotrypsin (ACT) deficiency
Serum concentrations of α1-antichymotrypsin for the three different −15 α1-antichymotrypsin signal peptide genotypes Ala/Ala, Thr/Thr, and Ala/Thr
Discussion
Proteinase-antiproteinase imbalance is an important cause of the chronic pulmonary disease in CF. Neutrophil elastase is found in excess in the lungs of CF patients and is thought to be a key mediator of lung destruction.3 ,4 Cathepsin G is also released by activated neutrophils and is similarly found in excess in the lungs of patients with CF.3 Its exact function is not known, but it has a bactericidal function against Gram negative organisms in vitro23 ,24 when uncontrolled cathepsin G can have deleterious effects on the lungs as it can digest connective tissue and augment the proteolytic effects of neutrophil elastase.23 ,25 ,26 Moreover, it stimulates airway gland secretion.27 Alpha1-antichymotrypsin is a serine proteinase inhibitor that can inhibit cathepsin G.28 Previous studies have shown an association between deficiency of this protein and COPD.16 ,17 One might therefore predict that CF patients with α1-antichymotrypsin deficiency would be more prone to tissue destruction from uncontrolled proteinase activity. However, we have found that patients with CF with a plasma deficiency of α1-antichymotrypsin had less severe pulmonary disease than those with normal levels of this protein.
These results support our previous observations that mild to moderate deficiency of the serine proteinase inhibitor α1-antitrypsin is associated with less severe pulmonary disease in patients with CF.5 ,6 Interestingly, patients with CF with α1-antichymotrypsin deficiency were also less likely to be colonised with P aeruginosa, which was independent of CF genotypes. Although there was no significant difference in the mean ages between the α1-antichymotrypsin deficient and normal groups, the deficient group had a wider standard deviation in their ages. This makes it possible that some of the negative correlation betweenP aeruginosa colonisation and α1-antichymotrypsin deficiency could result from the fact that the younger individuals with α1-antichymotrypsin deficiency had not yet contracted the organism. Nevertheless, the differences in percentage predicted FEV1 between the groups still remained significant when accounting for age in the analysis. Colonisation with P aeruginosa is known to be associated with more severe pulmonary disease in CF, but the effect of α1-antichymotrypsin deficiency on lung disease was independent of this factor. This suggests that the bactericidal effect of cathepsin G confers protection against colonisation withP aeruginosa in patients with α1-antichymotrypsin deficiency and CF.
Pulmonary inflammation is known to contribute to the pulmonary damage in CF. Alpha1-antichymotrypsin is an acute phase protein so it is possible that the low α1-antichymotrypsin levels could reflect a lesser degree of inflammation (which may be linked to less colonisation with P aeruginosa), and hence less severe pulmonary disease. However, mitigating against this is the fact that all of the deficient individuals had α1-antichymotrypsin levels below the lower limit for the normal reference range, despite the fact that most had evidence of an active inflammatory response. Although none of the patients with α1-antichymotrypsin deficiency had mutations in the signal peptide or coding sequence, they may have had a single nucleotide polymorphism in a regulatory sequence which could affect baseline or stressed levels of the protein as has been described for tumour necrosis factor (TNF)α.29
Alpha1-antichymotrypsin and cathepsin G both participate in the regulation of inflammation. Deficiency of α1-antichymotrypsin would lead to an excess of cathepsin G and decreased amounts of α1-antichymotrypsin-cathepsin G complex and cleaved α1-antichymotrypsin. The α1-antichymotrypsin-cathepsin G complex and cleaved α1-antichymotrypsin are chemotactic for neutrophils,30 and the complex stimulates the production of interleukin (IL)-6.31 Moreover, free cathepsin G can cleave IL-6 and the Fc regions of immunoglobulins. This would inhibit the IL-6 induced activation of neutrophils and reduce the ability of immune complexes to stimulate an oxidative burst in neutrophils.32 ,33 These are natural feedback mechanisms that serve to limit inflammation and may explain the less severe pulmonary phenotype in individuals with α1-antichymotrypsin deficiency.
Alpha1-antichymotrypsin is a major component of the filamentous plaques present in the brain of patients with Alzheimer's disease34 and it has been suggested that the –15 Ala/Ala signal peptide genotype increases plasma levels of α1-antichymotrypsin and so increases the risk of developing Alzheimer's disease.18 We assessed the effect of the –15 signal peptide genotype on the plasma levels of α1-antichymotrypsin, taking into account the degree of inflammation, and found that the mutation had no effect on the plasma levels of the proteinase inhibitor.
Previous studies have shown no difference in the frequencies of the –15 signal peptide genotype between patients with COPD and controls.16 We found an increased incidence of the –15 Ala allele in patients with CF but no link with α1-antichymotrypsin deficiency. Furthermore, patients with the –15Ala/Ala signal peptide genotype had a better percentage predicted FEV1 and chest radiographic score in a multivariate analysis that was independent of α1-antichymotrypsin and α1-antitrypsin deficiency. This raises the possibility that the –15 Ala allele is linked with another protective factor. The small sample size makes it important to repeat these studies in a larger group of patients with CF.
In summary, we have shown that deficiency of the proteinase inhibitor α1-antichymotrypsin is associated with less severe pulmonary disease in patients with CF and with lower rates of colonisation with P aeruginosa. The relatively small numbers in the deficient group indicate that these results should be viewed as preliminary and larger studies are required. However, these data are consistent with our previous studies of α1-antitrypsin deficiency and support the hypothesis that the breakdown products of proteinase inhibitors, coupled with an increased concentration of free enzyme, can downregulate a harmful inflammatory response. Further studies evaluating the inflammatory and infective processes in the lungs of patients with proteinase inhibitor deficiency will provide insights into the role of these regulatory mechanisms in disease.
Acknowledgments
We are grateful to Dr Nick Carroll, Addenbrooke's Hospital, Cambridge for help with the chest radiographic scoring and Roger Westerbeek, MRC Biostatistics Unit, University of Cambridge for help with the statistical analysis. This work was supported by the Cystic Fibrosis Trust (UK).