Article Text

Statistics from Altmetric.com
The goal of this review is to describe barrier function of the intestine, the structure of the tight junction, methods to evaluate intestinal permeability, and most importantly the relevance of abnormal permeability to disease. In this context, we will also present an emerging paradigm regarding the genesis of autoimmune diseases and describe the data that supports this from the perspective of both human disease and animal models. While this is a complicated area there are several points worth remembering:
epithelial permeability of the gastrointestinal tract can be evaluated in a site specific manner;
increased intestinal permeability is observed in association with several autoimmune diseases. It is observed prior to disease and appears to be involved in disease pathogenesis;
there are new and novel therapies directed at altering abnormally increased intestinal permeability and these may play a role in treating or preventing these diseases.
BARRIER FUNCTION
From the lower oesophageal sphincter to the anus, the gastrointestinal tract has a single contiguous layer of cells that separates the inside of the body from the external environment. Separation is important as there are a wide variety of environmental agents in the lumen of the bowel that can initiate or perpetuate mucosal inflammation if they cross the epithelial barrier. While the epithelial lining of the intestine plays a critical role in preventing access of these agents, it is not the only component of what is termed barrier function. Also important are secreted products such as immunoglobulin, mucous, defensins, and other antimicrobial products.
The importance of epithelial barrier function in normal homeostasis can be appreciated from experiments performed in the early 1990s where cell wall extracts from luminal bacteria were injected into the colonic wall of rats.1 This simple manoeuvre of bypassing the epithelial barrier and placing luminal compounds directly into the colonic wall initiated an inflammatory disease that was characterised by granulomatous reactions both in the bowel and, importantly, as a systemic inflammatory process. Similar results were also observed by variably expressing a dominant negative N-cadherin mutant in epithelial cells along the crypt-villus axis. This induced abnormal function of intercellular junctions and initiated a mucosal inflammatory disorder that resembled Crohn’s disease.2 The important concept that emerges from these experiments is that by simply abrogating epithelial barrier function, inflammatory disease can be induced in a susceptible host with features that are expressed both locally and systemically.
ANATOMICAL STRUCTURE OF THE EPITHELIAL BARRIER
Much of the epithelial barrier is formed by the rigid lipid bilayer of the enterocyte brush border. As in most cell membranes, this structure has appreciable solubility to lipid compounds but offers a strong barrier to water soluble constituents. The enterocyte balances its dual function as both an absorptive and barrier cell by embedding transport systems within this membrane for the water soluble compounds that it wishes to transport. However, at the junction between epithelial cells there is a potential route for solute traffic that is not regulated by brush border membrane transporters or channels. In order to regulate traffic through this paracellular pathway, mammalian epithelial cells form a series of intercellular junctions along their lateral margins. Closest to the luminal surface lies the tight junction and underneath is the adherens junction.
These structures are enormously complex in both their lipid and protein constituents. An ever expanding family of proteins are found in the vicinity of these junctions, forming fibrils that cross the plasma membrane to interact with proteins from the adjoining cell. These proteins also interact intracellularly with the actomyosin ring that encircles the enterocyte at the level of the tight junction through numerous smaller proteins. The fibrils between cells consist of at least two types of tetraspanning membrane proteins, occludin and members of the claudin family. The latter is comprised of at least 19 different but related proteins, their name coming from the Latin “to close”. It is of interest that defects in claudins have already been associated with human disease: a genetic mutation in claudin 16 appears to be involved in renal hypomagnesaemia, characterised by massive renal loss of magnesium.3 Furthermore, claudins 3 and 4 are targets for the bacterially produced toxin, Clostridia perfringens enterotoxin which dramatically increases tight junctional permeability in tissue culture systems.4
On the intracellular side of the membrane, the carboxy terminal end of these proteins interacts with the tight junction proteins ZO-1, ZO-2, and ZO-3 (see fig 1⇓). These proteins belong to the membrane associated guanylate kinase superfamily and possess an enzymatically inactive guanylate kinase-like domain. Underneath the junctional complex lies a ring of actin microfilaments and contraction of this has been proposed to regulate paracellular permeability. Connecting this ring to the junctional complex (ZO family members) are a series of actin filaments, as schematically outlined in fig 1⇓. In addition to these protein constituents, there are numerous other junctional proteins that have been described and a tremendous effort is underway to elucidate the physiological interactions of these proteins in terms of cellular and junctional function. Excellent reviews of tight junctional structure and the protein composition of these fascinating organelles are available.5–,8 One emerging concept is that the relative abundance of the different claudin family members is important in determining the physiological properties of the junction. Along normal developmental axes (such as along the crypt-villus axis) or during disease expression, the relative abundance of the claudins can change by up to 1000-fold while other more structural protein components seem to change relatively little.9
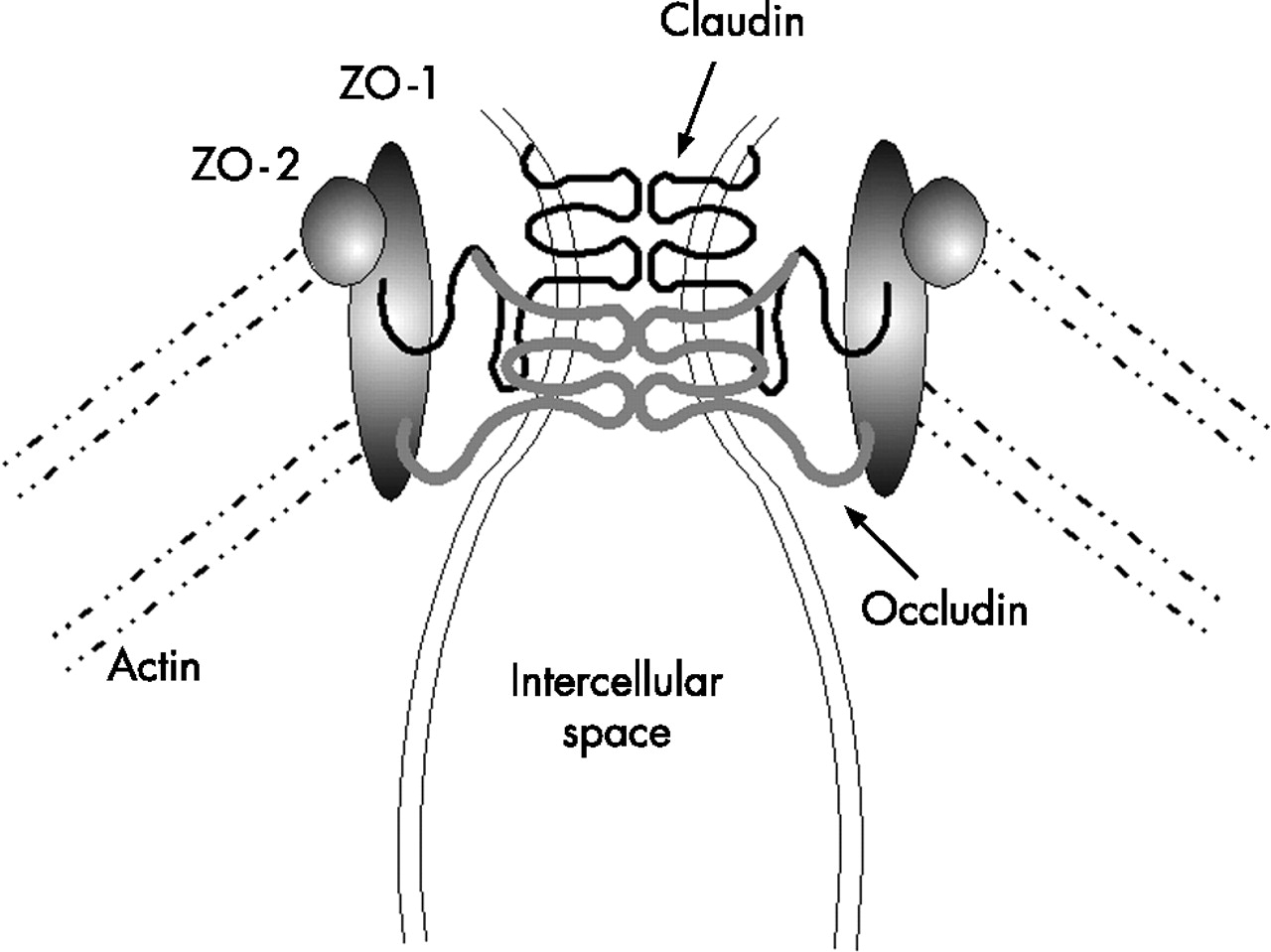
Tight junction structure. Homotypic association of claudin and occludin is illustrated. Numerous other proteins have been described and for a detailed description of their function and localisation, the reader is referred to the reviews given in the text.
It is also important to consider what factors alter permeability of the junction. We now recognise that the functional state of the tight junction, once considered a static parameter, is in reality incredibly dynamic. Epithelial tight junctions open and close all the time in response to a variety of stimuli. These include dietary state, humoral or neuronal signals, inflammatory mediators, mast cell products, and a variety of cellular pathways that can be usurped by microbial or viral pathogens.10–,19 A complete discussion of all of these mechanisms is beyond the scope of this article but a few deserve closer attention as they are important in our understanding of disease pathogenesis.
The first are dietary factors. It is now generally appreciated that the permeability of the paracellular pathway can be modulated by transcellular absorptive processes. During activation of the sodium dependent glucose transporter (SGLT1), there is a physiological opening of tight junctions that allows for the movement of small molecules and peptides.20–,23 This pathway will accommodate particulate sizes of the order of 2000 molecular weight (MW) but still exclude large particles such as horseradish peroxidase (MW ∼40 000).24 Although this is a normal physiological event, the purpose it subserves remains unclear. However, as discussed in the next section, this observation is important in our understanding of how to understand permeability measurements.
A physiological pathway, relevant to disease, is the zonulin pathway. Many bacteria alter tight junction state, presumably to enhance their own growth requirements. Vibrio cholerae secretes a variety of toxins and one of these, zonula occludens toxin, was recognised as increasing paracellular permeability. The mechanism by which this occurred was novel and involved binding to an apical membrane receptor on the enterocyte with subsequent activation of an intracellular pathway resulting in actomyosin contraction and increased paracellular permeability. The investigators speculated that it was unlikely that this pathway was present for the sole benefit of the bacteria and that a more likely scenario was that the pathway was a physiological one that bacteria had evolved to take advantage of. This proved to be true and in an elegant piece of work this bacterial toxin was used to identify the human homologue for this pathway now termed zonulin.25 It appears that in many scenarios where permeability is increased, a common pathophysiological event is upregulation of zonulin secretion from a lamina propria source into the lumen26–,28 with inappropriate activation of this pathway. The end result is increased paracellular permeability. This is illustrated in fig 2⇓.

Zonulin pathway. Depicted is a schematic view of what is known about the regulation of zonulin. Multiple steps are illustrated. (1) A luminal trigger for zonulin release arises and interacts with receptors, presumably on the epithelial cell. Triggers can be bacterial or food products such as gluten. (2) The trigger is sensed and a signal is passed to a lamina propria cell (3) where zonulin is released, diffuses to the luminal compartment, and thereafter binds to an apical receptor (4). This initiates a complex series of intracellular events that ultimately involves phosphorylation of tight junction proteins and opening of the paracellular space (5–6).
Measurement of permeability
Over the past several decades there has been a concerted effort to develop simple non-invasive means to evaluate the permeability properties of the paracellular pathway. In order to rationally evaluate methods to quantitatively evaluate the paracellular pathway in vivo, it is important to keep in mind a few simple principles. Movement across this pathway occurs by a non-carrier mediated process and as such depends on several features.
The concentration gradient across the barrier.
The surface area of the epithelium.
The time available for permeation.
The intrinsic permeability properties of the barrier.
As the last item is the characteristic of interest for measurement, it is important to try and avoid differences in the first three variables during the measurement process by carefully selecting the probes used and the method of their employment. Historically, a wide variety of probes have been utilised to determine paracellular permeability properties. Typically, they have several features in common; they are usually small, water soluble, non-charged compounds that are not destroyed in the gut, are non-toxic, not metabolised or sequestered once absorbed, and quantitatively cleared by the kidney into the urine. Finally, they should be easily detectable in urine and easily separated from similar endogenous or dietary compounds. This area has been extensively reviewed recently.29,30
Despite the proliferation of available probes, the majority of work in humans and experimental animals has employed a variety of small saccharide probes and/or Cr-EDTA. This is simply because these probes satisfy the criteria above, are cheap, and easily detectable. However, it is important to recognise that several of these probes are destroyed by processes that take place in the lumen of the gut and this limits their exposure to the epithelium in a manner that can be advantageously used to evaluate permeability characteristics in a regional manner.
As an example, sucrose is a useful probe for determining permeability characteristics of the gastroduodenal region.31 Distal to the gastroduodenal region, sucrose is rapidly hydrolysed by sucrase-isomaltase and therefore permeation of intact sucrose across the gastrointestinal mucosa must occur in the most proximal regions of the gut. In a similar manner, the traditional small intestinal permeability probes, lactulose, mannitol, rhamnose, or cellobiose, are degraded by the bacterial flora of the large intestine and yield no information regarding colonic permeability characteristics. Furthermore, under conditions of small intestinal bacterial overgrowth, loss of lactulose and mannitol is impossible to quantify and calls into question determination of permeability. In order to evaluate colonic permeability properties, probes must be selected that are stable in this environment. These include either Cr-EDTA or sucralose.32 Although these probes are stable throughout the gastrointestinal tract, they preferentially provide information regarding colonic permeability because, under normal conditions of intestinal transit, they reside within the colon for the majority of their time in the gut. The principles and techniques of probe selection for regional permeability determinations are discussed more extensively elsewhere32 but the basic features of these probes are schematically depicted in fig 3⇓.
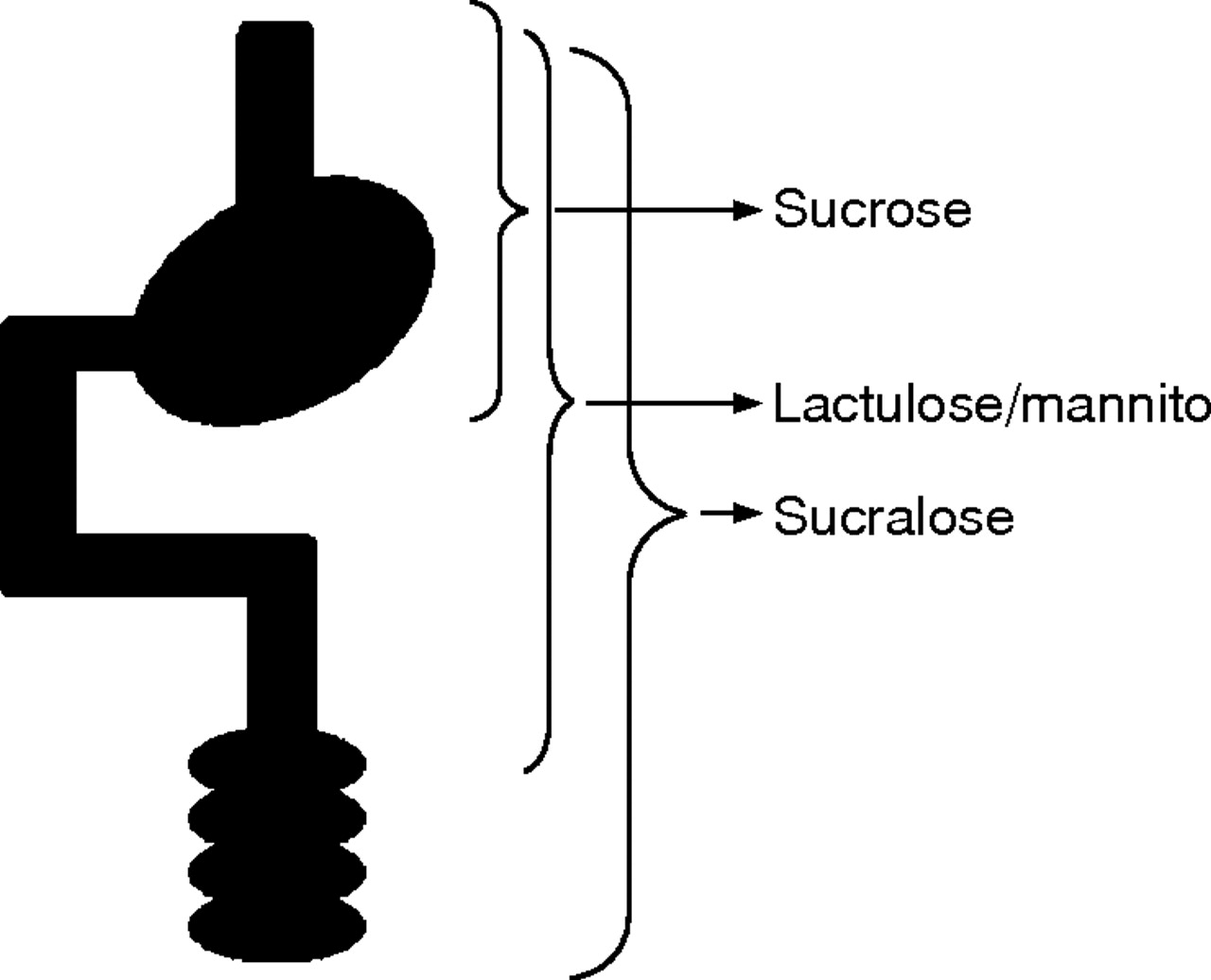
Region specific permeability measurements. By carefully choosing probes that have only a limited exposure to the gastrointestinal epithelium site, selective permeability determinations can be made. Sucrose is destroyed once it leaves the stomach and so sucrose permeability is a reflection of gastroduodenal disease. In a similar manner, lactulose and mannitol are destroyed in the caecum and provide information regarding the small intestinal epithelium. Finally, probes such as sucralose and Cr-EDTA are stable throughout the gut. They provide preferential information regarding the colonic epithelium as they spend most of a 24 hour exposure period in that organ.
Perhaps the most important issue to consider is what the permeation rate of the various probes actually means. The most concise hypothesis regarding the routes various probes take across the epithelium has been provided by Fihn and colleagues.33 These investigators provided data to suggest that there are a series of “aqueous pores” distributed along the crypt-villus axis of the small intestine. At the tips of the villus are relatively abundant small channels (radius <6 A) while in the crypts there exist much larger channels (50–60 A) in low abundance. At the base of the villus are intermediate sized (10–15 A) channels. The channels at the tips of the villus are increased in number by addition of glucose22 and susceptible to solvent drag effects while those in the crypt are unaffected by these alterations. The intermediate sized channels seem to be unaffected by solvent drag effects, perhaps as under physiological conditions this part of the villus is not exposed to luminal contents. With these data it would appear that, under normal conditions, molecules the size of disaccharides (for example, lactulose) are restricted from moving across the villus tip whereas mannitol can do so with relative freedom. This hypothesis is illustrated schematically in fig 4⇓ and provides one possible explanation for the marked difference in permeation rates for probes with small differences in physical size.

Small intestinal permeation pathways. Depicted is a schematic representation of the crypt-villus axis. The broken line is an estimated depth of “luminal exposure”—the limit to which solvent drag effects are noticeable. This limit is loosely based on the known distribution of the sodium dependent glucose transporter 1 along this axis. Within this region glucose stimulated water flux is evident and can also drag particles that can fit through the same pathways as water is using. On the left is a qualitative estimate of increasing pore size along the crypt-villus axis with a horizontal bar providing a qualitative estimate of the number of each sized pore. The location of uptake for the various sized probes is shown on the right with the number in parentheses referring to the hydrodynamic radius of each probe in angstroms.
Traditionally, small intestinal permeability is expressed as the ratio of the fractional excretion of a larger molecule to that of a smaller one (for example, lactulose:mannitol). Using the paradigm from the preceding paragraph, this would represent the number of intermediate sized pathways as a proportion of the total number of aqueous channels. As the smaller channels are concentrated at the villus tips, permeation rates of compounds across this pathway become a rough assessment of mature small intestinal surface area. This is well established from a clinical perspective. In diseases where there is a marked reduction in mature small intestinal surface area, such as coeliac disease, there is a substantial reduction in the fractional excretion of small probes such as mannitol. Coupled with this there is an increase in the fractional excretion rates of larger probes such as lactulose. This would suggest either an increased number of intermediate sized channels per unit surface area or the appearance of an alternate pathway, accessible to lactulose, that is not evident under normal physiological circumstances. As intermediate sized channels appear to be present in the immature parts of the crypt-villus axis, relative expansion of this crypt-villus fraction (as observed in coeliac disease) is a possible explanation. However, there are also data demonstrating that the presence of either epithelial damage or increased rates of apoptosis provide alternate routes for the permeation of larger molecules such as lactulose.34–,36
The net result of a reduction in the fractional excretion of mannitol and an increase in that of lactulose is a dramatic increase in the lactulose:mannitol ratio. The converse is seen during the healing process of the coeliac lesion. During treatment with a gluten free diet, the first sign of recovery is seen as an increase in the fractional excretion of mannitol, suggesting that recovery of mature surface epithelium precedes the reduction in apoptosis and damage.37 This interpretation fits well with the known pathophysiology of coeliac disease.
Therefore, a reasonable interpretation of permeability data, expressed as a lactulose:mannitol ratio, is the amount of epithelial damage as a proportion of mature small intestinal surface area. It is important to remember that this parameter provides no information about either colonic or gastric permeability. The former is due to the fact that both probes are destroyed in the colon and the latter since the surface area of the small intestine is so much greater than that of the stomach. In order to obtain this type of information, additional probes such as sucrose and sucralose are necessary and provide the necessary data without detracting from the measurement of small intestinal permeability. Additional probes to assess absorptive function can also be added to extend the usefulness of such testing.38,39
NEW PARADIGM FOR AUTOIMMUNE DISEASE
As discussed previously, there are data available suggesting that abrogation of epithelial barrier function can induce inflammatory disease, either locally or remotely. We will offer a hypothesis to explain this observation and then provide experimental data to support this. In order to do this we will consider type 1 diabetes as a model disease and then extend these observations to several other diseases that we believe have similar aetiologies.
We hypothesise that many diseases have a pathogenesis that consists of three main features, represented diagrammatically in fig 5⇓.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disease initiation pathway. Depicted is a proposed model of the initiation of autoimmune disease. In this model, an initiating factor is found in the environment (lumen of the gut) that is kept separate from the mucosal immune system by a competent epithelial barrier. Abrogation of this barrier by any mechanism allows for initiation of the disease process.
A genetically susceptible immune system (the mucosal immune system), that allows the host to react abnormally to an environmental antigen.
An environmental product that triggers the disease process.
The ability for the environmental agent to interact with the mucosal immune system. Since the purpose of the epithelial barrier is to keep these two factors separate, and we measure this function of the barrier by permeability, the corollary of this is that an increase in permeability is a requirement for disease expression.
Within this model the specificity for disease location (target tissue) is provided by both the antigen and the genetic abnormality of the immune system. For instance, the target may be the beta cells of the pancreatic islets (diabetes), the epithelial cells of the gut (coeliac disease), or the myelin sheaths surrounding nerve (multiple sclerosis). In some cases the antigen is known, such as gluten for coeliac disease, but for most it is not and may represent a dietary antigen, an antigen from the host flora, or a transient infectious antigen. The antigen may cross the epithelial barrier at any level of the gut and this may provide some clues to the type of antigen involved. As we will demonstrate, permeability alterations can be observed at several levels of the gut and at each of these the type of prevalent antigen differs. For instance, in the proximal gut, intact dietary antigens will be common but in the distal gut, antigens from the microflora will be more abundant. This model also does not place any requirements on how the increase in permeability arises. This can occur during an infectious process, by activation of endogenous humoral pathways, or by microbial manipulation of the host’s epithelial cell pathways. It may also be a transient event and this may be an explanation for the lack of detectable permeability abnormalities in some patients.
So, what is the evidence to support this paradigm? Probably the best example is that of type 1 diabetes in the BB rat. In this model of autoimmune diabetes an inbred line of rat develops classical autoimmune diabetes when weaned onto a normal diet.40 However, if the animal is instead weaned onto a hydrolysed diet, a much lower incidence of diabetes results. This is an inbred line of animals and so a genetic susceptibility of the mucosal immune system is not an unreasonable proposition. However, the observation that simply altering the diet can dramatically change the risk of disease suggests a dietary trigger that can be removed by protein hydrolysis. In view of the paradigm outlined above, we would also predict that these animals should have increased intestinal permeability. When measured, there is a marked increase in permeability of both the small intestine (lactulose/mannitol ratio) and of the stomach (sucrose).41 The increase in permeability was not due to diet as animals receiving the hydrolysed diet had the same increase as those on the regular diet.
The increase in permeability is not present at all ages and so the question arises as to what is the mechanism of the increased permeability. As discussed previously, one potentially abnormal pathway is the zonulin system to open the paracellular pathway. Recent work has demonstrated that in the BB rat, coincident with the increase in permeability, there is increased zonulin secretion into the lumen.42 Therefore, in this model of autoimmune disease we now have evidence for a dietary antigen and an abnormality of the zonulin pathway that leads to increased permeability. The disease can be prevented by removing the antigen, but most exciting of all, it can also be prevented by abolishing the increased permeability. This was recently demonstrated by Watts et al using a zonulin receptor antagonist which prevented the increase in small intestinal permeability and expression of diabetes.42 The message inherent in these experiments is that expression of diabetes requires the genetic predisposition of the rat strain, the dietary provocative agent, and abnormal permeability. Removal of either the luminal antigen or the permeability defect prevents disease, despite retaining the genetic predisposition.
HUMAN DISEASE
There are a variety of human diseases in which it has been suggested that abnormal permeability is important. These include: diabetes, Crohn’s disease, coeliac disease, multiple sclerosis, irritable bowel syndrome, and a pot pourri of others. Each will be discussed in the following sections.
Diabetes
Given that the BB rat model of type 1 diabetes provides strong support for increased permeability playing a key role in disease pathogenesis, it is perhaps not surprising that this has been further investigated. In the non-obese diabetic mouse there have not been any studies evaluating alterations in permeability but there is evidence of a coeliac-like enteropathy with an increased number of intraepithelial lymphocytes.43 In other situations with these findings, increased permeability is typically present and it would not be a surprise if it were present in this mouse model of type 1 diabetes.
In humans, increases in intestinal permeability in type 1 diabetics were first reported 20 years ago.44 These early studies did not exclude patients with concomitant coeliac disease nor did they use some of the more modern detection methods for the probes. However, more recent studies, that have taken these into account, continue to find that type 1 diabetic patients have increased small intestinal permeability.45,46 This is not simply related to hyperglycaemia or poor glycaemic control as patients with type 2 diabetes have normal permeability.47
Of great interest is a recent publication48 that examined 339 type 1 diabetic patients and 89 of their first degree relatives. In this work it was demonstrated that diabetic patients had significantly higher serum zonulin levels than either controls or their relatives. Furthermore, by examining serum stored for HLA testing, these authors also demonstrated that “pre-diabetics” (positive for IA2 autoantibodies but before the onset of diabetes) had elevated zonulin levels in seven of 10 specimens. Patients with increased zonulin, not unexpectedly, also had increased permeability.
Therefore, the data seem fairly convincing that in both animal models and human type 1 diabetes, a substantial percentage of individuals have increased permeability that appears to precede the disease state and may play a role in pathogenesis.
Crohn’s disease
The aetiology of Crohn’s disease remains unknown. However, the three features discussed above appear important. Environmental factors are critical; the disease is unlikely to develop in susceptible animal models under germ free conditions49 or in humans where luminal contents are diverted. From these data it would appear that luminal antigens or other factors are critical. A genetic component is also apparent and several gene mutations have been identified, including those in NOD2/CARD1550 as well as in the organic cation transporter (OCTN or IBD5).51,52
In animal models of Crohn’s disease, the interleukin 10−/− mouse, the SAMP mouse, and the mdra deficient mouse, it has now been convincingly demonstrated that abnormal permeability is present prior to expression of the inflammatory disease.53–,55 It is unclear whether preventing the increase in permeability will prevent disease as these experiments have not yet been done but the observation of increased permeability prior to development of disease is now well established. In some models, the permeability increase is present months before disease expression.54
In the human condition it is also evident that increased small intestinal permeability is commonly observed in populations at high risk of developing Crohn’s disease.56–,61 Increased permeability is observed in the absence of symptoms of disease, suggesting that it is not merely an early manifestation of Crohn’s disease. There is now one case report of a young woman found to have abnormal permeability during a family study who subsequently developed Crohn’s disease.62 At the time when her permeability was increased, extensive investigations revealed no evidence of bowel disease. It took years for Crohn’s disease to develop. There are also data to suggest that patients with increased permeability are also likely to have evidence of subclinical inflammation, as assessed by calprotectin excretion.63
There is also an apparent link between increased permeability and the recently described mutation in the NOD2/CARD15 gene. In a recent study, Buhner et al demonstrated that not only is the risk of having abnormal permeability and a NOD2 mutation increased among first degree relatives of those with Crohn’s disease, but these are related. Relatives with a NOD2 mutation had a 75% chance of having increased permeability,64 suggesting that this mutation may influence epithelial permeability. The mechanism by which this might occur is unclear. The NOD2 gene product does not appear to be present in enterocytes where the permeability abnormality is expressed. However, the gene product is present in Paneth cells and there the mutation is associated with a reduction in secretion of defensins,65 an observation previously reported to be associated with Crohn’s disease.66 This may represent an important link as reduced defensin secretion would be expected to alter the luminal microflora which in turn can have profound effects on epithelial permeability by many different mechanisms.67–,69
While an altered microflora, either induced by altered Paneth cell secretion or other mechanism, can directly increase permeability, there are other potential aetiologies. Altered expression of tight junction proteins may also increase paracellular permeability. In fact, increased expression of claudin-2 has been demonstrated to increase permeability,70 presumably as homodimers of this protein form associations that are not as “tight” as heterodimers with other claudins. In this regard, it is of interest to note that upregulation of claudin 2 expression has been reported in patients with Crohn’s disease and ulcerative colitis.71 Whether this is an effect of the inflammation itself or a pre-existing abnormality could not be determined. However, other authors have reported that the abnormality in expression of junctional proteins is specific to biopsies taken from inflamed regions of ulcerative colitis and Crohn’s disease patients; those portions of the gut that were not inflamed appeared to be similar to the control biopsies in terms of junctional proteins.72
In terms of function, Soderholm and colleagues73 reported that baseline permeability in non-inflamed portions of gut taken from patients with Crohn’s disease is similar to controls, but following exposure of the tissues to a mildly damaging agent (sodium caprate) the non-inflamed tissue demonstrated a marked increase in paracellular permeability. This would suggest that there is a subtle alteration of function, independent of inflammation, which can be manifest as an increase in paracellular permeability. Similar findings are observed in vivo manifesting as hyperresponsiveness of the small intestine to the damaging effects of non-steroidal anti-inflammatory compounds such as aspirin or ibuprofen.74,75
Taken together, these data support the hypothesis that there is a permeability abnormality in Crohn’s disease and that this abnormality is important in the genesis of disease. At least some of the reported genetic associations of Crohn’s disease appear to involve these pathways.
Coeliac disease
Coeliac disease also appears to follow the paradigm. It is an exceedingly common disease that may affect up to 1% of the population and has clear genetic and environmental components. From a genetic perspective, almost all coeliac patients (95%) are HLA-DQ2 positive while the remainder carry HLA-DQ8.76 There is also evidence of other important genetic linkages.77 With this disease we have the good fortune to have identified the inciting environmental antigen—gluten. Removal of this agent prompts complete remission of all attributes of the disease, including a return of abnormal intestinal permeability to almost the normal range in the majority of subjects.78 In fact, an increase in permeability is a sensitive test for the presence of even small amounts of gluten in the diet.78 However, if intestinal permeability normalises during disease remission, how can there be any evidence that increased permeability plays a role in the aetiology of this disease?
In fact, there is increasing evidence that this is the case. The first line of evidence comes from animal studies. An inbred Irish Setter dog line develops a gluten sensitive enteropathy that mimics human coeliac disease. In these animals the disease can be completely prevented by weaning the animal onto a gluten free diet. However, subsequent exposure to the antigen immediately prompts development of the disease. Importantly, animals that have never been exposed to dietary gluten have increased small intestinal permeability.79 This strongly suggests that in this animal model abnormal permeability precedes disease.
In humans the data are more difficult to interpret as we do not have good data in individuals prior to the onset of disease. However, with careful analysis it is apparent that tight junctional structure is abnormal in children with coeliac disease.80,81 In some studies gluten removal does not totally resolve the defect, suggesting that either some portion of the damage is irreversible or that there is a prior alteration in the tight junction.81
From a functional viewpoint, intestinal permeability has not been well assessed in relatives of patients with coeliac disease. In one study about one third of first degree relatives had abnormal permeability.82 Approximately 8% of these had a positive endomysial antibody test, underwent biopsy, and were demonstrated to have asymptomatic coeliac disease but the reason for the abnormal permeability in the remainder was not reported.
Patients with dermatitis herpetiformis (DH) provide an interesting perspective in this regard. Subjects with this condition exhibit an enormous range of associated bowel pathology from frank coeliac disease to a completely normal intestinal biopsy and no evidence of bowel disease. In a recent study of 18 such patients28 it was noted that all DH patients had increased intestinal permeability, including those patients without evidence of intestinal disease. Furthermore, these patients also had elevated serum zonulin levels, suggesting that the abnormal permeability was associated with, and perhaps caused by, an abnormality of the zonulin pathway. As some of these patients may go on to develop coeliac disease, it would appear that in these cases increased permeability would precede development of disease and a potential mechanism of this is upregulation of the zonulin pathway.
The association of abnormal permeability and increased zonulin secretion in a gluten sensitive disease (DH) is intriguing. Recent work suggests that in coeliac disease, gluten functions as a trigger for zonulin release which subsequently increases paracellular permeability.83 These events were found to be MyD88 dependent suggesting the involvement of a TLR pathway that did not appear to be either TLR2 or TLR4. The authors suggest a model of coeliac disease in which gluten first interacts with the innate immune system through a TLR pathway. This subsequently induces release of zonulin from a lamina propria source, increased paracellular movement of gluten, and enhanced interaction of gluten with the mucosal immune system initiating the inflammatory disease process. This model is a minor variation on the paradigm presented.
Other diseases
There are a variety of other diseases in which a similar disease paradigm may be at work. Atopic dermatitis has been suggested to involve dietary antigens and many patients describe a dietary association to disease flares. However, convincing evidence of abnormal permeability in this patient group is hard to find. Many studies that have examined relatively small numbers of patients found no evidence of altered permeability84–,86 while several others have found abnormalities in a subgroup of patients.87–,89 In at least some of these patients relief of symptoms has accompanied probiotic treatment90 and in one study probiotics not only reduced the skin disease but also decreased elevated small intestinal permeability.91 It is unclear from this study whether the increased permeability was a cause of the atopic dermatitis or an associated area of inflammation. From the data available it is difficult to conclude whether atopic dermatitis fits the proposed disease paradigm.
Rheumatological conditions have long been associated with abnormalities of intestinal function and the concept of abnormal reactivity to a luminal antigen in these conditions is prevalent. Perhaps the best evidence for this comes from the literature on ankylosing spondylitis. Increased gastrointestinal permeability has been recognised to be present in these patients for decades but it was unclear whether this was due to the disease or its treatment with non-steroidal anti-inflammatory drugs (NSAIDs).92 With more recent work, the effect of NSAIDs has been isolated and it is apparent that these patients appear to have a primary defect in intestinal permeability that is also shared by a subgroup of relatives.93 In this study, the permeability defect was limited to the small intestine and not expressed in the stomach. However, in contrast, a recent study has suggested that patients with ankylosing spondylitis and Helicobacter pylori infection not surprisingly have increased gastric permeability.94 When the bacteria was eradicated, increased gastric permeability to sucrose fell in non-arthritic patients but remained elevated in those suffering from spondylitis. The authors suggest that this represents a pre-existing increase in gastric permeability, perhaps important in disease pathogenesis.
Irritable bowel syndrome (IBS) is also interesting to consider in this regard. In the paediatric population there is a recognised association between the symptoms of IBS, food hypersensitivity, and in some cases true food allergy. When intestinal permeability was measured pre and post provocation with suspect foods, it was demonstrated that these patients developed abnormal permeability.95 However, the concept of IBS developing as a consequence of, or in concert with, a functional derangement of gut function is best exemplified by the observation that this condition is commonly observed following acute infectious gastroenteritis. The strongest association is with Campylobacter infections and in this condition increased permeability can be observed up to a year following infection.96 In another large study involving IBS following a waterborne outbreak of gastroenteritis, increased permeability was more commonly observed in patients with IBS than controls.97 These studies provide intriguing data that abnormal permeability may be involved in disease pathogenesis, although they cannot exclude an alternative explanation that mild gut damage (measured by increased permeability) is associated with the development of IBS by another mechanism. Hopefully, the ability to interfere therapeutically with pathways that regulate intestinal permeability will allow us to establish whether increased permeability is a necessary prerequisite for the development of these diseases.
CONCLUSIONS
The paracellular pathway between intestinal epithelial cells has become important in our understanding of gastrointestinal and systemic disease. Long thought to be a static non-regulated barrier to the passage of luminal material, it is now recognised to be a dynamic constantly changing structure with a functional state that is carefully regulated. Luminal organisms can modulate the state of the tight junction through multiple mechanisms and while opening tight junctions may be of benefit for the microflora, it may be deleterious to the host. Abnormal function of this pathway can also be observed in conditions where the structure of the proteins comprising the junction is abnormal.
The functional state of the junction can be assessed by measuring the rate of movement of probes across the junction. This is what is referred to as gastrointestinal permeability testing and there are a variety of means to accomplish this either in vivo or in vitro. By carefully selecting the method used, it is possible to measure the permeability of various sites within the gastrointestinal tract.
For decades a variety of pathological states have been associated with abnormal permeability. Many of these are a consequence of intestinal epithelial damage that is associated with disease but not involved in a causal manner in the genesis of disease. However, in several autoimmune conditions it appears that increased permeability is a constant and early feature of the disease process. Furthermore, it is becoming increasingly apparent that in some conditions increased permeability is critical to the development of disease as if it is abrogated the disease does not develop. This is particularly true in type 1 diabetes. In other diseases such as Crohn’s disease or coeliac disease, a similar pattern of findings are apparent but the experiment to try and prevent disease by preventing the increase in permeability has not been performed.
REFERENCES
Supplementary materials
Competing interest: JBM is a member of the scientific advisory board of ALBA Pharmaceuticals. There are no other competing interests.