Article Text

Statistics from Altmetric.com
The role of stress in the modulation of the most common gastrointestinal disorders has traditionally been considered a domain of psychology, and has frequently been lumped together with the role of psychiatric comorbidity. Among clinicians, the term “stress” is generally taken as synonymous with psychological (“exteroceptive”) stress. Based on the deeply ingrained Cartesian view in medicine and gastroenterology, stress and psychological factors have been considered fundamentally separate and unrelated to the “real” biological changes underlying organic disease. However, recent breakthroughs in the understanding of the neurobiology of the organism's response to acute and chronic stress, and the evolving understanding of elaborate brain-gut interactions and their modulation in health and disease, are beginning to require a reassessment of chronic stress in the pathophysiology and management not only of functional but also of “organic” gastrointestinal disorders.
Certain stressful life events have been associated with the onset or symptom exacerbation in some of the most common chronic disorders of the digestive system, including functional gastrointestinal disorders (FGD), inflammatory bowel disease (IBD), gastro-oesophageal reflux disease (GORD), and peptic ulcer disease (PUD). Even though methodological differences in reported studies which do and do not support such an association remain to be resolved, the association of sustained stressful life events preceding symptom exacerbation is based on several well designed surveys in patients with FGD,1-4with post-infectious irritable bowel syndrome (IBS),4 and with IBD.5-8 In addition, acute life threatening stress episodes in adult life (rape, post-traumatic stress syndrome) are an important risk factor in the development of functional gastrointestinal disorders.9 Finally, early life stress in the form of abuse plays a major role in the susceptibility of individuals to develop functional as well as IBD10-14 later in life. Thus, depending on the type of stressor, the lag time between the stressful event and the clinical manifestation or exacerbation of FGD or IBD may range from decades to weeks.
Even though the recent focus on Helicobacter pylori in the aetiology of PUD has nearly abolished the interest in the role of stress in PUD, there is considerable evidence that supports a role of stressful life events in the aetiology of PUD.15-21 Furthermore, more than 80% ofH pylori infected individuals (and the majority of non-steroidal anti-inflammatory drug (NSAID) users) never develop an ulcer, while at least 10% of non-NSAID related peptic ulcers are not infected with H pylori.22 It is intriguing to speculate on the role of certain life stressors as risk factors which determines whichH pylori positive individual actually develops an ulcer and which patients develop symptoms of dyspepsia instead, without an ulcer.
In contrast with FGD, IBD, and PUD, the epidemiological evidence to support a causal relationship between life events and disease activity in GORD is less conclusive. The primary information on the role of stressful life events is based on a population based survey in which 64% of patients with GORD indicated that stress increased their symptoms.23 GORD patients who are anxious and are exposed to long periods of stress are more likely to notice stress induced symptom exacerbation.24
Stress response: defence of homeostasis at the cost of allostasis
Stress, defined as acute threats to the homeostasis of an organism,25-27 be they real (physical) or perceived (psychological), and whether posed by events in the outside world or from within, evokes adaptive responses which serve to defend the stability of the internal environment and to assure the survival of the organism.28 Surprisingly, despite the wide range of different types of stressors, some of the principal circuits underlying the stress response under these different circumstances are remarkably similar.28 However, while the pathways involved in the activation of hypothalamic effector neurones duringinteroceptive stressors (gut infection, mucosal inflammation, internal haemorrhage) may be conceived as simple reflex responses, mediated at a subcortical level by the system involved in the processing of visceral information,exteroceptive stressors (psychological) engage circuits in the limbic forebrain, including the lateral and medial prefrontal cortex, hippocampus, and amygdala.28Involvement of cortical circuits plays an important role in adjusting the stress response to the context, the physiological state of the organism, memories of past stressful life events, and beliefs about the subjective meaning of the situation.
Elaborate neurobiological response systems have evolved to orchestrate an integrated response which is best suited to respond to a specific stressor in a given situation for a specific individual. This ability to defend homeostasis (that is, to maintain stability) through change has been referred to as allostasis.29 In the healthy individual, the physiological response systems are rapidly turned on and off, synchronising the physiological stress response to the duration of the stressor, and limiting the exposure time of the organism to the potentially harmful effects of the stress response. However, there are several situations in which the severity or chronicity of the stressor and the ensuing physiological response systems can cause damage, exacerbate existing disease processes, or predispose the individual to acquire new diseases—that is, become maladaptive. This is particularly true in situations where the responsiveness of physiological responses to stress and the ability to adapt has already been altered due to genetic30 or early life events,31 thereby biasing an individual's susceptibility to the negative effects of stress throughout life. These long term effects of the organism's accommodation to certain types of stress have been referred to as allostatic load,32 the “wear and tear” resulting from chronic overactivity or underactivity of physiological stress response systems. Stressors which have been associated with such maladaptive consequences, both acute and chronic, are referred to in this review aspathological stressors. The outcome of pathological stress on the patient is determined not only by the length, severity, and type of stressor, but also by other factors, such as genetics, early life experiences, cognitive factors, and environmental support.
Physical and psychological stressors
Systemic or interoceptive stressors,28 in the context of the four chronic gastrointestinal disorders listed above, can occur in the form of mucosal inflammation (IBD, PUD) or tissue irritation by excessive acid exposure (GORD). Inflammatory cytokines, including tumour necrosis factor α (TNF-α), interleukin (IL)-1, and IL-6 cause acute stimulation of the hypothalamic-pituitary-adrenal (HPA) axis alone or in synergy.33 ,34 The cytokine mediated stimulation is mediated through stimulation of corticotropin releasing hormone (CRH; or corticotropin factor, CRF) and arginine vasopressin release from hypothalamic neurones, and by direct effects at the pituitary and adrenocortical levels. Different mechanisms, including cytokine stimulation of vagal afferents, have been proposed by which the cytokine signal crosses the blood-brain barrier.28 The ultimate output of peripheral cytokine stimulated HPA axis activation, plasma cortisol, is the principal negative feedback mediator to shut off both the inflammatory response as well as HPA axis activation. In contrast with the effects of acute inflammation, several animal experimental34 ,35 and human studies in patients with certain chronic inflammatory disorders, such as rheumatoid arthritis, have provided evidence for a blunting of the HPA axis response.34 ,36-38 This blunting appears to be secondary to downregulation of CRH gene expression and CRH secretion by mediators associated with chronic inflammation.
Psychological or exteroceptive stressors fall into different categories, depending on the individual's age during stress exposure, severity and chronicity of the stressor, and the subjectively perceived threat. For example, several different types of psychological stressor can have permanent consequences on the responsiveness of the individual to stress and chronic disease later in life: (1) Early life stress in the form of altered mother-infant interaction during a species specific “window” of development has been shown to result in permanent hypersecretion of CRF and overactivity of the locus coeruleus.31 (2)Chronic abuse (physical or sexual) and neglect throughout life have been found to be associated with alterations in the HPA axis response to stress.39-41 (3) Exposure to a one time stressor which is perceived by the individual as life threatening (rape, combat situation, natural disaster) resulting in post-traumatic stress syndrome42 with alterations in sympathetic and HPA axis responses to stress and to exaggerated memory recall of the traumatic event.43 Chronic stressors in adult life (such as losses, financial threats, etc), in particular when sustained and perceived as threatening, may result in transient reversible alterations in allostatic systems, which in the case of IBS have been shown to result in exacerbation of IBS symptoms.3 It is primarily these latter types of pathological stressors that have been addressed in the epidemiological studies mentioned earlier.
How are stressors translated into integrative physiological responses?
While the traditional concept of stress has focused on subjective conscious feelings, thoughts, beliefs, and memories reported by some individuals in association with stressful life events, the major breakthroughs in this area have occurred through an understanding of the biological mechanisms which are responsible for the detrimental effect of certain stressful life events on health. The focus of this review is on these biological mechanisms and their implications, while the reader is referred to the extensive cognitive literature.44-48
The organism's response to stress is generated by a network comprised of integrative brain structures, in particular subregions of the hypothalamus (paraventricular nucleus, PVN), amygdala, and periaqueductal grey. These structures receive input from visceral and somatic afferents and from cortical structures. Cortical inputs include the medial prefrontal cortex, and subregions of the anterior cingulate and insular cortices.28 ,49 ,50 In turn the integrative network provides outputs to the pituitary and pontomedullary nuclei, which in turn mediate the neuroendocrine and autonomic output to the body, respectively.28 ,46 ,49 This central stress circuitry is under feedback control via ascending monoaminergic projections from these brain stem nuclei, in particular serotonergic (raphe nuclei) and noradrenergic (including locus coeruleus) nuclei, and via circulating glucocorticoids (GCs), which exert an inhibitory control via central GC receptors located in the medial prefrontal cortex and hippocampus.34 The parallel outputs of this central circuitry (“emotional motor system”)51 which is activated in response to various stressors include responses of the autonomic nervous system, the HPA axis response, the endogenous pain modulation system, and ascending aminergic pathways. These pathways are summarised in fig 1.
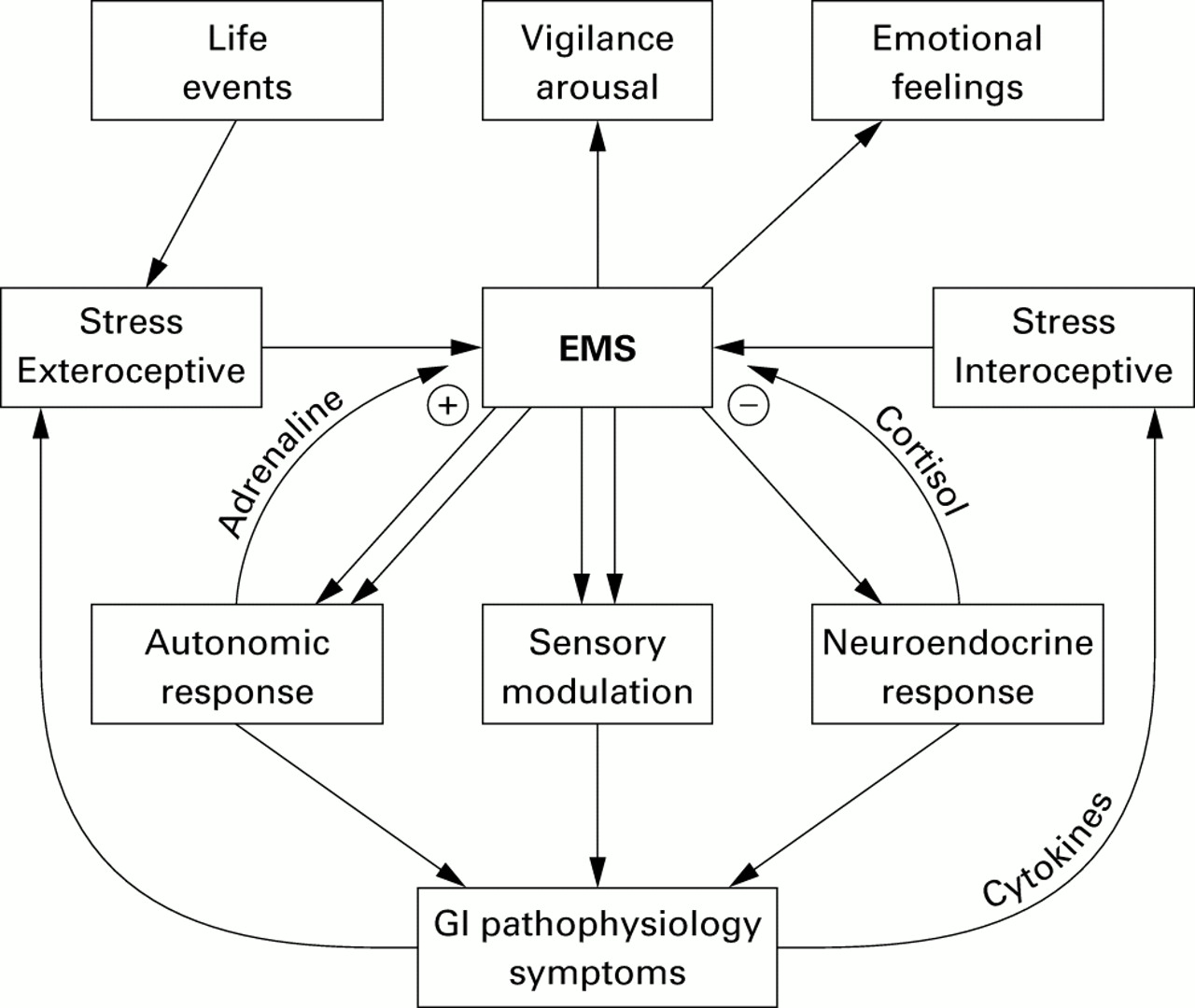
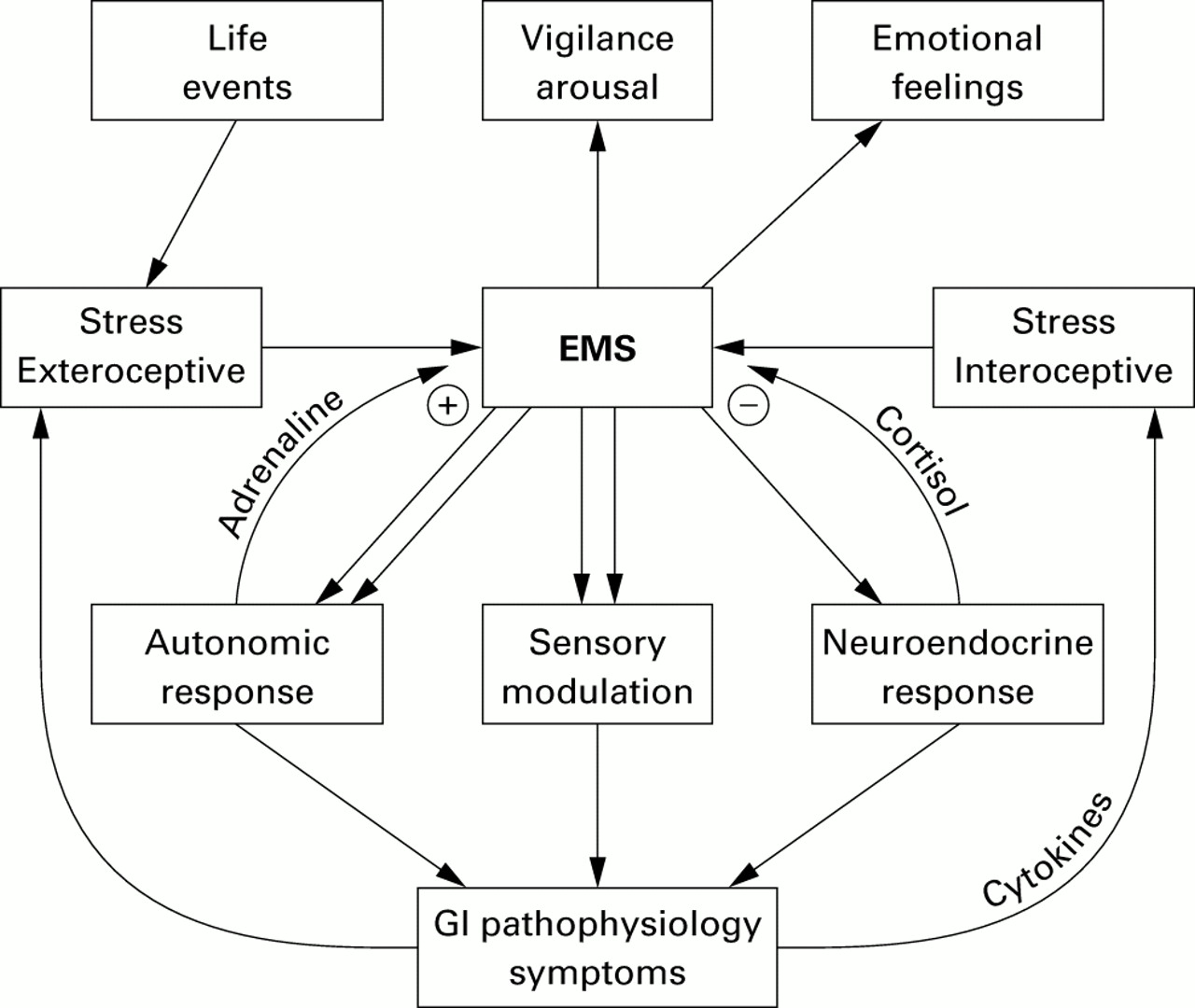{kind=link}
Emotional motor system (EMS) pathways.
One important chemical mediator of the central stress response is CRH (and probably related, currently unknown molecules) located in certain effector neurones of the PVN, the amygdala, and locus coeruleus complex.52 CRH secretion by PVN neurones is under positive feedback regulation by central noradrenergic pathways (including those originating from the locus coeruleus), thereby forming a bidirectional positive feedback loop between the CRH-noradrenergic systems.53 ,54 Central injection of CRH can reproduce behavioural and physiological responses similar to those seen in response to acute psychological stress,55 and inhibition of CRF mediated responses by antagonists56 ,57 or in knockout animals results in a decrease in the animal's response to stress.58 ,59
Modulation of physiological stress responses by allostasis
The responsiveness and output pattern of this network is likely to be under partial genetic control30 ,60 and shows considerable plasticity in response to early life events31and to certain types of pathological stress.61 ,62 For example, studies in animals and humans have clearly demonstrated that certain types of pathological stress can alter the responsiveness of feedback systems by downregulation of pre and/or postsynaptic receptors (adrenergic, serotonergic, GC receptors)61 ,62 and in the most severe forms by structural changes in certain brain regions.63 ,64 Thus pathological stress can not only activate, but also fundamentally change, the responsiveness and output of the central stress circuits. These alterations could affect the individual output pathways of the general stress response differentially and in different directions; for example, increase or decrease in target specific sympathetic outputs, increase or decrease in certain vagal outputs, up or downregulation of the HPA axis, and up or downregulation of pain perception. Some of the best characterised alterations in this central adaptation to pathological stress are an increase in CRF synthesis and secretion,65 ,66 ,67 an increase in the activity and sensitivity of central noradrenergic systems,31 ,68-71 and either downregulation or sensitisation of GC receptors and adrenocorticotropic hormone release.72 As a consequence of these alterations in the central stress circuitry, secondary changes in receptor systems can occur in spinal73 or peripheral target cells of the output systems.74 Thus in cases of pathological stress resulting in permanent changes in the central stress circuitry, life long changes in peripheral receptor systems may also be expected. Finally, changes in mood and affect associated with alterations in the stress response have been reported.32 ,75
AUTONOMIC RESPONSE
The classical description of the autonomic nervous system to stress in the context of the “fight and flight” response by Cannon76 has focused on the stereotypic and global activation of the sympathetic nervous system. However, despite the integrated nature of the response to different stressors, there is considerable variability in the specifics of the peripheral output. At the level of the PVN of the hypothalamus, the cells that give rise to major classes of visceromotor projections are separate from another, suggesting they are not necessarily called into play in a stereotyped all or none matter, but rather that the potential exists for differential recruitment.77 In the periphery, 12 different functional groups of sympathetic neurones have been identified.78 Some of these pathways regulate mucin production by large intestinal goblet cells, net water absorption by intestinal epithelial cells,79 mucosal permeability,80 ,81 mast cell degranulation,82 and possibly release of peptides from enterochromaffin cells.
Importantly, some of the sympathetic pathways have a direct immunomodulatory function.83 Evidence has been provided to suggest that there is a functionally distinct branch of the sympathetic nervous system dedicated specifically to immune modulation.78 Noradrenergic sympathetic nerve fibres innervate the vasculature and parenchyma of lymphoid organs, including the gut.84 These nerves and their principal neurotransmitter noradrenaline can influence (a) basic immune cell function such as proliferation, differentiation, cell trafficking, and cytokine production; (b) acquired immune responses, and (c) autoimmune reactivity in susceptible strains.83 For example, activation of the sympathetic system causes systemic secretion of IL-6 from immune cells. IL-6, by inhibiting TNF-α and IL-1β, and by activating the HPA axis, participates in the stress induced suppression of the immune-inflammatory reactions.85 The effects of the sympathetic system on immune cell function are consistent with the reported suppressing effect of naturalistic and certain experimental induced stressors on immune function.86 However, in experimentally induced urethral inflam- mation,87arthritis,88 and colitis89 in rats, sympathectomy has been shown to reduce inflammation.
Stress related increases in plasma adrenaline (and GCs) play an important role in the facilitation of memory in amygdala-hippocampal circuits, including the development of conditioned fear.90 Adrenaline stimulated vagal feedback has also been implicated in the activation of endogenous pain modulation circuits.91
In addition to activation of sympathetic pathways, variousacute stressors produce a characteristic biphasic pattern of parasympathetic activation, consisting of gastrovagal inhibition and activation of sacral parasympathetic output.92 Similar to the different subclasses of central sympathetic neurones, subpopulations of function and target specific vagal motor neurones have been identified. Pathological stress has also been associated with persistent decreases in cardiovagal tone and in cardiovagal responsiveness to stress.93 ,94
Viewed together, these results demonstrate that the interface between the gut lumen and neural, endocrine, and immune pathways is under close control of the autonomic nervous system. Alterations in the autonomic regulation of this interface, under conditions of allostatic load, are likely to play an important role in the modulation of secretion, motility, inflammation, and sensory response of the gut to luminal contents.
HYPOTHALAMIC-PITUITARY-ADRENAL (HPA) AXIS
The HPA axis is acutely activated by both interoceptive34 ,53 and exteroceptive stressors.75 The peripheral GC response to stress, in parallel with the sympathetic response plays a prominent role in suppression of the inflammatory response. In addition, a central mechanism in the normal counterregulation of stress induced HPA axis activation is GC mediated feedback by GC receptors in certain brain regions such as the hippocampus and medial prefrontal cortex (see fig1).95 ,96 This feedback mechanism observed in response to acute stress can be up or downregulated in various chronic disease states. Hyperactivity of the HPA axis manifesting as hypercortisolism as seen in certain forms of depression has been considered the classical form of a generalised stress response which has escaped its usual counterregulation.75 Such hyperactivity has also been reported in anorexia nervosa,97 panic disorder,98 and sexual abuse.41 A decrease in GC receptor expression has been observed in animal models of chronic stress99 and in adult animals exposed to perinatal stress.31 A decrease in central GC receptors may be secondary to reversible downregulation of the receptor or permanent destruction of GC containing brain regions.63 ,100 ,101
A different pattern of stress induced HPA dysregulation has been described in patients with post-traumatic stress disorder,72 chronic fatigue syndrome,102fibromyalgia,103 and possibly diarrhoea predominant IBS.104 Published reports suggest that these patient populations have evidence of a highly sensitised HPA axis characterised by decreased basal cortisol levels, increased number of lymphocyte GC receptors, greater suppression of cortisol to dexamethasone, and a more sensitised pituitary gland.72 Thus in addition to the classic pattern of increased cortisol levels in response to acute stress, there appears to be a pattern characterised by diminished cortisol levels as a result of a stronger negative feedback inhibition in certain types of disorders associated with pathological stress. This diminished cortisol response may be associated with increased central CRF responses to stress.65 On the other hand, decreased corticosterone response to stress has been reported in chronically stressed rats, associated with decreased expression of CRF mRNA in the PVN.105 The different patterns of HPA axis dysregulation may be related to genetic factors and/or develop in response to different types of pathological stress (for example, chronicity, severity).
In summary, HPA axis responses vary in different disease states with different impacts on duration of the stress response and on peripheral and central cortisol levels. Chronically elevated cortisol levels can be associated with structural irreversible changes in certain brain regions.
MODULATION OF GUT IMMUNE FUNCTION
Peripheral outputs of the stress response, in particular GCs and catecholamines, have profound effects on cytokine networks, including those in the gut mucosa.105a Via its peripheral mediators, stress influences the production of key regulatory type 1 and type 2 cytokines, T helper (Th) 1 and Th2 functions, and components of cellular and humoral immunity. In the healthy organism, both GCs and catecholamines suppress Th1 responses and cellular immunity and shift the immune response towards Th2 responses and humoral immunity. In contrast, in Crohn's disease, the response pattern is shifted towards Th1 responses.105b Based on these observations, one might speculate that the different patterns of the stress response in chronic functional and inflammatory conditions of the gut (see below) may have opposite effects on the Th1/Th2 balance in the gut mucosa.
MODULATION OF VISCEROSOMATIC SENSITIVITY
Both clinical and animal experimental data strongly support the concept of stress and fear induced analgesia resulting in decreased somatic pain perception.106 ,107 Stress induced analgesia is mediated by descending pain inhibitory pathways and, depending on the nature and severity of the stressor, is partially mediated by opioidergic, glutaminergic, and serotonergic systems.108Recent evidence suggests that this stress induced somatic hypoalgesia can be accompanied by a stress induced visceral hyper- algesia.109 ,110 One way to explain these observations is the concept that both pain facilitatory and inhibitory systems are activated simultaneously in response to stress, the net effect being determined by the relative contribution of these opposing influences.111 Another hypothesis assumes that the visceral hyperalgesia is indirectly mediated by a population of sympathetic and/or parasympathetic nerves, stimulating the release of chemicals from cells within the gut wall (for example, mast cells,112 enterochromaffin cells) with sensitising effect on visceral afferent terminals.112 The role of pathological stress on the relative activation of this dual activation has not been characterised.
In summary, stress is associated with modulation of visceral and somatic sensitivity. Alterations in these endogenous stress activated pain modulation systems may play an important role in several functional visceral or somatic syndromes characterised by chronic discomfort and pain.
MONOAMINERGIC SYSTEMS
Noradrenergic, serotonergic, and cholinergic projection neurones to cortical (prefrontal cortex) and subcortical (including PVN, amygdala, hippocampus, nucleus tractus solitarius) regions play an important role in emotional arousal and in feedback modulation of the emotional motor system. Evidence for reciprocal positive feedback regulation of hypothalamic CRF neurones and noradrenergic locus coeruleus neurones has been reported.54 ,113 For example, upregulation of tyrosine hydroxylase in locus coeruleus neurones has been reported in perinatally stressed rats which show upregulation of hypothalamic CRH,31 and parallel downregulation of message for CRH and tyrosine hydroxylase has been demonstrated during long term treatment of rats with imipramine.34 For example, noradrenergic modulation of synaptic vagal transmission in the nucleus tractus solitarius can be assumed to play a role in the modulation of vagovagal reflexes,62 ,114 ,115 including those regulating gastric accommodation, regulation of spontaneous transient lower oesophageal sphincter relaxation, and duodenogastric reflexes. Chronic overactivity of these systems, as observed in certain animal models of social stress62 and in patients with post-traumatic stress syndrome68 has been shown to be associated with downregulation of autoreceptors (α2, 5-HT1Areceptors)62 ,116 resulting in enhanced release of noradrenaline and serotonin, respectively. Enhanced release in turn may result in downregulation of postsynaptic receptors (such as β adrenergic and α1 receptors).62 ,117 These neuroplastic changes would initially increase noradrenaline release (presynaptic) but ultimately decrease activation of postsynaptic target neurones. In addition, excessive release of transmitter could result in depletion of noradrenaline and serotonin further decreasing postsynaptic neurone activation. The time course and extent of these adaptive changes to chronic stress differ between involved brain regions.62 ,116 Thus similar to alterations in the GC mediated system, neuroplastic alterations in aminergic systems may play a prominent role in the chronic biasing of the stress response towards maladaptive responses.
In summary, central aminergic networks involving serotonergic, noradrenergic, and cholinergic pathways play an important role in mediating the output of the central stress response both to specific regions of the brain (arousal, emotion) and to the periphery (autonomic, pain modulation). Alterations in the gain and effectiveness of these networks are likely to play a central role in the wide range of maladaptive responses to pathological stress, manifesting as both affective and somatic disorders.
Possible role of allostasis in chronic gastrointestinal disorders
The association of disease activity with certain types of stressors in FGD, IBD, and probably GORD suggests that the alterations in specific outputs of the central stress circuits, as well as the adaptive changes in peripheral target cells induced by pathological stress and facilitated by genetic factors, play a pathophysiological role in disease activity. While such altered outputs may be solely responsible for predominant symptoms in IBS and possibly functional dyspepsia, they may only play a modulatory role in the other disorders. It is important to realise that fear conditioning and interoceptive conditioning118 are likely to play important roles in triggering stress responses to situations and contexts which by themselves are not threatening or stressful.119 Below we will summarise selected reports of altered autonomic, neuroendocrine, and endogenous sensory modulation in FGD, IBD, and GORD, viewing these alterations as changes not in isolation but as alterations in the integrated response to stressors as described in fig 1.
Functional gastrointestinal disorders
CHANGES IN AUTONOMIC NERVOUS SYSTEM RESPONSES
In both healthy humans and animals, stressors have been shown to result in a characteristic stress induced slowing of gastric emptying,120 increase in distal colonic motility,121 ,122 and acceleration of intestinal transit.92 ,123 In the most common functional gastrointestinal disorders, IBS and FD, persistent alterations of autonomic responsiveness is likely to play a role in altered bowel habits and alteration in gastric emptying, respectively. Evidence for such alterations in IBS includes increased responses of distal colonic motility in response to laboratory stress121 and possibly food intake124 and delayed gastric emptying in a subset of patients.125
A model of IBS, taking into account altered autonomic regulation of gastric and distal colonic function and based on upregulation of CRF containing neurones in Barrington's nucleus (part of the locus coeruleus complex) has recently been reported by Valentino and coworkers.52 While descending CRF containing projections from this pontine nucleus to the distal colon may mediate increased stress and food induced motor responses of the distal colon, ascending projections to the locus coeruleus and to the forebrain may be responsible for mediating arousal and shifting attention to visceral afferent stimuli. Increased expression and release of CRF in IBS patients, or a subset of patients, are also consistent with the reported evidence for increased sympathetic responses.104 ,126-128
Changes in the frequency of high amplitude propagated contraction in the colon, presumably via alteration in vagal colonic regulation, may play an important role in diarrhoea and slow transit constipation, thereby determining the predominant bowel habit pattern in IBS.129-131 In FD, reported findings of delayed gastric emptying, decreased antral motor activity to food and stress,120 ,132 and impaired proximal gastric accommodation to a meal133 and to duodenal distension134 are also consistent with an alteration in the gain of vago-vagal reflexes, possibly by central monaminergic systems, as discussed above.
There is evidence that decreased cardiovagal tone is present in certain patients with functional dyspepsia135 ,136 and in subsets of patients with IBS.136 ,137 Recent evidence from patients with functional constipation suggests that despite the heterogeneity of vagal motor neurones, changes in cardiovagal tone, vagal regulation of intestinal transit, and vagal regulation of colonic mucosal blood flow may all be reduced in parallel.138
HPA AXIS CHANGES
Evidence for alterations in HPA axis function has been demonstrated in diarrhoea predominant IBS patients who showed decreased 24 hour plasma cortisol, blunted cortisol responses, and normal adrenocorticotropic hormone responses to noxious rectosigmoid distension.104 In contrast, Heitkemperet al reported that urine cortisol levels obtained immediately on rising were significantly higher in IBS women compared with control women.128 Even though a thorough characterisation of HPA axis responses in FGD patients has not been reported, these preliminary findings suggest the pattern of sensitised GC feedback also reported in victims of abuse,39fibromyalgia, and chronic fatigue syndrome.139 There is significant overlap in the epidemiology of all of these conditions with IBS.10 ,140-143 While it is currently not known if these HPA axis changes are an epiphenomenon or play a role in symptom generation and pathophysiology of these syndromes, one may speculate on their possible role in the observed findings in post-infectious IBS patients. The reported persistence of chronic inflammatory mucosal changes after eradication of the infectious organism,4 and increased intestinal permeability and hyperplasia of enterochromaffin cells144 are consistent with an inadequate physiological response to acute gut inflammation, in particular an inadequate cortisol (and possibly an altered sympathetic) response. One may speculate that a downregulated cortisol response to intero- and exteroceptive stressors might also predispose IBS patients to chronic inflammatory conditions, such as asthma, rheumatoid arthritis, or IBD.145
CHANGES IN PAIN MODULATION
Suggestive evidence for alterations in stress induced modulation of viscerosomatic sensitivity comes from human and animal studies. IBS patients show cutaneous hypoalgesia146 ,147 combined with visceral hypersensitivity,148 a similar pattern as seen in the rat in response to psychological stressors.110Preliminary results using psychological laboratory stress in healthy volunteers suggests a stress induced increase in colonic or rectosigmoid sensitivity to distension.109 Even though all published human studies are open to methodological criticism, they are consistent with reported findings in animals of differential viscerosomatic pain modulation. It is of interest to note that patients with bulimia (who, in contrast with IBS patients have a hyperactive HPA axis) show cutaneous hypoalgesia also which precedes symptom exacerbation.149
CHANGES IN BRAIN ACTIVATION
Functional brain imaging studies of IBS patients have shown decreased activation of the perigenual cingulate and hippocampus.150 Decreased perigenual cingulate/medial prefrontal cortex activity has also been reported in patients with depression151 and with post-traumatic stress disorder.152 Bremner et alreported a decrease in prefrontal and orbitofrontal cortical metabolism in patients with post-traumatic stress disorder in response to the α2 antagonist yohimbine.68 Together with results from preclinical studies showing decreased metabolism in cortical regions with high noradrenaline release,153 these results are consistent with enhanced noradrenaline release in these brain regions in patients with post-traumatic stress disorder. One may speculate that the decreased activation in the perigenual cortex and other brain regions seen in IBS patients may also be related to exaggerated noradrenaline release in response to stress.
In summary, IBS patients, in particular the non-constipated subpopulation, present with a pattern consistent with enhanced stress responsiveness manifested by predicted autonomic and pain modulatory responses, and sensitised GC feedback. This response pattern is associated with changes in brain activity in response to stress consistent with increased central noradrenaline release. The blunting of the HPA axis may precede the onset of IBS symptoms and may predispose individuals to develop post-infectious IBS. The fact that up to 40% of IBS patients show evidence of increased anxiety,154 and the fact that the changes are similar to those reported in a variety of other so called “functional” disorders, suggest a top down model, in which the alterations in the central stress circuits in predisposed individuals are triggered by pathological exteroceptive stressors and play a primary role in pathophysiology.
Inflammatory bowel disease
CHANGES IN AUTONOMIC NERVOUS SYSTEM RESPONSES
Little is known about specific alterations in the autonomic nervous system responses to interoceptive or exteroceptive stressors in patients with IBD or in animal models of colitis. However, several animal studies provide indirect evidence for involvement of autonomic dysregulation.
Qiu and colleagues155 recently provided evidence that short term moderate stress can enhance the response of the colon to chemically induced inflammation. Their findings are consistent with a model in which the effect of the stressor is primarily mediated by autonomic responses, and not by stress induced alterations in neuroendocrine function (HPA axis). The authors show that CD4+ cells which are sensitised by a chemically induced colitis can be reactivated by a subthreshold dose of the same chemical irritant applied to the colon six weeks after the initial insult, at a time of complete mucosal healing. They provide evidence that this effect requires sensitised CD4+ lymphocytes, and is mediated in part by an effect of the stressor on mucin production and colon permeability, presumably facilitating access of the irritant to the sensitised lymphocytes. A previous study by the same group in the trinitrobenezene sulphonic acid rat model had demonstrated stress induced increases in myeloperoxidase levels in the rat colon, six weeks after induction of colitis.156 A series of articles from Perdue's group provided evidence for stress induced increases in mucosal permeability of the rat intestine.81 ,82 ,157Pharmacological evidence for mediation of these permeability changes by cholinergic and adrenergic nerves, mast cell degranulation, and involvement of peripheral CRH has been provided.81 The source of peripheral CRF in these stress mediated changes is not known but may include immune cells, postganglionic sympathetic neurones, and colonic enterochromaffin cells. In this context, it is of interest that increased intestinal permeability has also been reported in certain patients with post-infectious IBS symptoms.144
Taken together, these findings in animals together with the extensive literature on immune modulation by the sympathetic nervous system indirectly support a role for stress mediated activation of certain sympathetic (in addition to parasympathetic) nerves in increasing the permeability of the gut, altering the quantity of mucin, and altering immune function in the reactivation of inflammatory mucosal changes in chronic colitis. As activation of sympathetic nerve pathways during acute stress is generally associated with an immunosuppressive role, alterations in stress activated autonomic output, possibly associated with altered cortisol responses, must be responsible for a maladaptive response.
CHANGES IN HPA AXIS RESPONSES
Little is known about HPA axis regulation in patients with IBD. One may speculate that similar to patients with rheumatoid arthritis,36 HPA axis responses are downregulated by the chronic colonic inflammation in IBD patients, thereby compromising the organism's ability to counterregulate mucosal inflammation.158 Preliminary studies in rats with TNB induced colitis are indeed consistent with such downregulation (Tache, personal communication, 1999). Furthermore, in analogy to findings in Fisher and Lewis rats34 there may be genetic factors, such as a hyporesponsive HPA axis in subsets of IBD patients, that predispose to the persistence of inflammation. Evidence for such greater susceptibility of CRF hyposecreting Lewis rats to stress induced colitis has recently been provided.92 However, no evidence for a role of alterations in stress induced HPA axis activation has been found between rodents with short term colitis that showed stress induced recurrence of mucosal changes and those that did not.155 ,156 ,159 ,160 As only single time point measurements of corticosterone responses to stress were reported in these studies, it is not known if diurnal variation or 24 hour corticosterone output was altered in these animals.
In summary, one can only speculate that IBD patients may show similar downregulation of the HPA axis as patients with rheumatoid arthritis. However, in contrast with IBS, fibromyalgia, or post-traumatic stress disorder patients where this blunting appears to be related to increased central CRH release combined with enhanced GC receptor mediated feedback, HPA axis downregulation is likely to be secondary to decreased CRH gene expression and secretion.
CHANGES IN PAIN MODULATION
The limited information on visceral and somatic pain perception in patients with IBD suggests that acute inflammatory changes (at the site of distension) are associated with enhanced visceral sensitivity,161 while patients in remission have normal perception of rectal distension.162 Patients with Crohn's disease of the small bowel show normal or decreased sensitivity to rectal distension.163 In contrast, induction of colonic pain by repetitive sigmoid stimulation resulted in a decrease in rectal pain sensitivity, consistent with adequate activation of endogenous pain inhibitory pathways.164
CHANGES IN BRAIN ACTIVATION
Preliminary results comparing regional brain activation assessed by O15 water positron emission tomography between healthy control subjects, and patients with mild ulcerative colitis and IBS suggest similar activation patterns both during rectal distension as well as during anticipation of such distension, while IBS patients showed decreased activation of the perigenual cingulate, amygdala, and hippocampus.165
In summary, our understanding of the outputs of the central stress circuits in IBD is incomplete. However, it appears to be different from that observed in FGD patients. One may speculate that the output is comprised of downregulation of the HPA axis, alteration of gut targeted branches of the sympathetic nervous system, and normal activation of stress induced pain modulation pathways. In contrast with the pattern seen in IBS patients, the pattern in IBD may reflect genetic predisposition to a hyporeactive HPA axis combined with secondary downregulation in the central response to a chronic interoceptive stressor (for example, mucosal inflammation). In those IBD patients who appear to have comorbid IBS, the low cortisol response may be associated with alterations in autonomic and pain modulation seen in IBS patients.
GORD
Despite the suggestive epidemiological data on the association of GORD with stressful life events, little is known about possible alterations in the peripheral outputs of the central stress system in subgroups of patients with GORD. Recent evidence is consistent with a primary role of spontaneous transient relaxations of the lower oesophageal sphinter in the mediation of pathological acid reflux.166 One may speculate on the possible role of altered transmission of vago-vagal reflexes by central aminergic systems in this dysregulation (see above). Alteration of the gastro-oesophageal high pressure zone by changes in diaphragmatic function may also contribute to stress induced symptom exacerbation. Changes in diaphragmatic function related to stress induced breathing patterns have been reported.167 In addition, slowing of gastric emptying by inhibition of vagal gastric regulation may contribute to stress induced symptoms. Finally, recent evidence suggests a role for endogenous pain modulation systems in stress induced increase in oesophageal chemosensitivity.168
In conclusion, recent breakthroughs in understanding the neurobiology of stress emphasise the importance of the peripheral outputs of the central stress response in the modulation of some of the most common gastrointestinal disorders. The same central alterations discussed above may also explain the association with affective disorders seen in a certain percentage of patients with functional gastrointestinal disorders, and the overlap with a variety of clinical syndromes such as fibromyalgia, chronic fatigue syndrome, and interstitial cystitis. Animal models of different pathological interoceptive and exteroceptive stressors9 ,31 ,64 ,169 ,170 are available and, together with knockout technology will help to determine which components of the altered stress response are epiphenomena and which play a primary role in the pathophysiology. Finally, a better understanding of the role of pathological stressors in modulation of disease activity will have important therapeutic implications. Both pharmacological interventions (for example, CRF1 antagonist) as well as educational-behavioural interventions in selected patient populations are likely to become an integral part of cost effective disease management of FGD, IBD, and GORD.
Abbreviations used in this paper
- FGD
- functional gastrointestinal disorders
- IBD
- inflammatory bowel disease
- GORD
- gastro-oesophageal reflux disease
- PUD
- peptic ulcer disease
- IBS
- irritable bowel syndrome
- NSAID
- non-steroidal anti-inflammatory drug
- TNF-α
- tumour necrosis factor α
- IL
- interleukin
- HPA
- hypothalamic-pituitary-adrenal
- CRH
- corticotropin releasing hormone
- CRF
- corticotropin releasing factor
- PVN
- paraventricular nucleus
- GCs
- glucocorticoids
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 105a.↵
- 105b.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.↵
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.↵
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.↵
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.↵
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.↵
- 168.↵
- 169.↵
- 170.↵