Article Text

Statistics from Altmetric.com
Autoimmune hepatitis (AIH) is characterised by female predominance, interface hepatitis, hypergammaglobulinaemia, and autoantibodies.1 ,2 Typically, the disease responds well to corticosteroid therapy, and clinical, laboratory, and histological remission can be achieved in 65% of patients within 18 months.3 ,4 Among Caucasoid Northern Europeans, its mean annual incidence is 1.9 per 100 000, and its point prevalence is 16.9 per 100 000.5 In the USA, AIH affects 100 000–200 000 individuals.6 It accounts for 2.6% of the liver transplants in the European Liver Transplant Registry7 and 5.9% of liver transplants in the USA.8
Classical autoimmune hepatitis
Three types of AIH have been proposed based on immunoserological findings but only two types have mutually exclusive autoantibodies and different clinical profiles.9 Type 1 AIH is the most common form of the disease worldwide and is associated with antinuclear antibodies (ANA) and/or smooth muscle antibodies (SMA). It affects all age groups and is associated with human leucocyte antigen (HLA) DR3 (DRB1*0301) and DR4 (DRB1*0401) in Caucasoid Northern European and North American patients.10 ,11 DRB1*0301 andDRB1*0401 influence disease expression and behaviour as well as susceptibility. Caucasoid patients with type 1 AIH and DRB1*0301 are younger, and have a higher frequency of treatment failure,12 relapse after drug withdrawal,13 and requirement for liver transplantation than patients with other alleles.14 In contrast, patients with DRB1*0401 are typically older, frequently have concurrent autoimmune diseases, and respond better to corticosteroids than counterparts withDRB1*0301.15
Type 2 AIH affects mainly children and is characterised by antibodies to liver/kidney microsome type 1 (anti-LKM1).16 The target autoantigen of type 2 AIH is the cytochrome mono-oxygenase P450 IID6 (CYP2D6),17 ,18 and five antigenic sites located between peptides 193–212, 257–269, 321–351, 373–389, and 410–429 are recognised by anti-LKM1.19 ,20 The main antigenic site is the peptide sequence 257–269 and the second most recognised sequence is 321–351. Most patients with type 2 AIH have anti-LKM1 that react against one of these short linear sequences,19 ,20 and this reactivity distinguishes them from those patients with anti-LKM1 and chronic hepatitis C.21 ,22 Susceptibility to type 2 AIH appears to relate toDRB1*0701,23 ,24 but HLA B14, DR3, and C4A-QO have also been incriminated.25 Immune diseases (especially vitiligo, autoimmune thyroiditis, and insulin dependent diabetes mellitus) and organ specific autoantibodies (antibodies to thyroid, parietal cell, and/or islets of Langerhans) occur commonly.16 Initial studies that suggested a poorer outcome in type 2 than in type 1 AIH16 have not been substantiated, and both type 1 and type 2 disease respond comparably to corticosteroids.26 ,27 Both types can have an acute, even fulminant, presentation, and prompt recognition and treatment are necessary to control inflammatory activity.28-30
AIH in association with anti-LKM1 has also been described in 15% of patients with autoimmune polyglandular syndrome type 1 (APS1).31-33 This syndrome is caused by a single gene mutation located on chromosome 21q22.3.34 ,35 The APS1 gene encodes for a transcription factor called the autoimmune regulator which is expressed in epithelial and dendritic cells within the thymus where it may regulate clonal deletion of autoreactive T cells and affect self tolerance. Ectodermal dystrophy, mucocutaneous candidiasis, multiple endocrine gland failure (parathyroids, adrenals, ovaries), autoantibody production, and AIH in various syndromatic combinations are features of the disease.36 Unlike other autoimmune diseases, APS1 has a mendelian pattern of inheritance, complete penetrance of the gene, no class II HLA associations, and no female predominance. The cytochromes CYP1A2 and CYP2A6 are the major hepatic autoantigens of APS1, and reactivities against these targets have distinguished patients with APS1 from those with type 2 AIH.36 ,37 Recent studies however in patients with anti-LKM1, AIH, and associated endocrinopathies have blurred this distinction by indicating anti-LKM1 reactivity against P450 IID6 (CYP2D6).38
Type 3 AIH is the least established form and has been characterised by antibodies to soluble liver antigen (anti-SLA).39Recently, anti-SLA have been shown to be identical to antibodies to liver-pancreas (anti-LP), and the autoantibodies are now designated anti-SLA/LP.40-42 Reactivities to cytokeratins 8 and 18 were described initially,43 and then glutathione S-transferases were proposed as autoantigens.44 A 50 kDa liver cytosolic protein is now considered the more likely target,45 and this antigen may be a transfer RNA complex involved in the incorporation of selenocysteine into polypeptide chains (tRNP(Ser)Sec complex).46 Antibodies to the tRNP(Ser)Sec complex have characterised patients with severe AIH and poor response to corticosteroids.47Reactivity to conformational epitopes of the 50 kDa tRNA associated antigenic protein has been demonstrated in patients with type 1 AIH, type 2 AIH, and ANA/SMA positive sclerosing cholangitis.48Patients with anti-SLA/LP have clinical and laboratory features that are indistinguishable from patients with type 1 AIH and they respond well to corticosteroids.42 ,49 The evolving experience with anti-SLA/LP indicates that they are additional markers of autoimmune hepatitis rather than hallmarks of a separate entity. Indeed, the original promoters of a type 3 AIH have had serious doubts about its existence.4 The major clinical value of testing for anti-SLA/LP may be in the reclassification of patients with cryptogenic chronic hepatitis.42 ,49
Diagnostic criteria
The diagnostic criteria for AIH have been codified and updated by an international panel.50 ,51 Adefinite diagnosis requires exclusion of viral, drug induced, alcoholic, and hereditary liver disease. Laboratory features must demonstrate substantial immunoreactivity and liver tissue must disclose at least portal mononuclear cell infiltration and interface hepatitis. The six month requirement to establish chronicity has been waived, and cholestatic clinical, laboratory, and histological changes preclude the diagnosis. Aprobable diagnosis of AIH requires the same histological findings as for definite disease but there may be less immunoreactivity and some exposure to potentially hepatotoxic drugs or alcohol.50 ,51 Furthermore, the probable diagnosis can be supported by demonstration of antibodies to asialoglycoprotein receptor, liver specific cytosolic antigen type 1, SLA/LP, and/or perinuclear antineutrophil cytoplasm.50-53
A scoring system can facilitate the diagnosis in patients with mixed manifestations, and it provides a mechanism by which to assess the strength of the diagnosis in different populations.50 ,51By weighing each component of the syndrome, discrepant features can be accommodated and biases associated with isolated inconsistencies avoided. The original scoring system has been validated,54and an unintended application has been to use it to assess the resemblance of various chronic liver diseases to classical AIH in a quantitative and objective fashion.55 ,56
Variant syndromes
Patients with features of AIH and another liver disease (“overlap syndromes”) or with findings that are inconsistent with the definite diagnosis of AIH (“outlier syndromes”) constitute the variant syndromes.56-60 These variant forms have been reported extensively but the overall experience with these conditions remains relatively small and anecdotal. Standardised diagnostic criteria have not been promulgated; experiences between institutions have not been compared; natural history for each variant form remains uncertain; and treatment algorithms have not been validated. Application of the scoring system proposed by the International Autoimmune Hepatitis Group has been useful in providing a template for diagnosis.55 ,56 ,59 This method however has not been validated for sensitivity, and its major value has been to estimate the frequency of occurrence in a systematic fashion. Eighteen per cent of patients with autoimmune liver disease have features that vary from the classical syndromes of AIH, primary biliary cirrhosis (PBC), and primary sclerosing cholangitis (PSC),56 ,59 and patients with PSC most commonly have concurrent features of AIH that confound the diagnosis.55 ,56 ,59 Recognition of these variants is important because they are common; their inclusion in classical diagnostic categories can distort perceptions of disease behaviour and outcome; responsiveness to conventional therapies may vary; and they may provide clues to the pathogenesis of the typical disorders.61
AIH AND PBC
AIH and PBC are generally easy to discriminate on the basis of clinical, laboratory, and histological findings. Patients with AIH however may have antimitochondrial antibodies (AMA), including those against the PBC specific M2 antigens (up to 8% occurrence).49 ,62 Consequently, the scoring system for AIH was adjusted to allocate greater weight against PBC by increasing the negative deduction for AMA seropositivity from −1 to −4.51
Histological features of cholangitis, including destructive cholangitis, may also be present in AIH,63-65 and copper stains of hepatic tissue may be positive and indicative of chronic cholestasis.62 The scoring system for AIH was adjusted further to accommodate these observations by increasing the negative deduction for histological evidence of bile duct injury from −1 to −3.51
The presence of AMA seropositivity and cholestatic clinical, laboratory, and/or histological features in patients with AIH indicates the overlap syndrome of AIH and PBC (table 1). Retrospective analyses indicate that this variant occurs in 5% of patients with AIH and in 19% of patients with PBC.56 ,59 Disease behaviour and treatment response depend mainly on the component of the AIH-PBC overlap that predominates. Patients with mostly AIH have high serum aspartate aminotransferase levels, serum alkaline phosphatase concentrations less than twofold normal, moderate to severe interface hepatitis on histological examination, and high diagnostic scores for AIH (aggregate scores ⩾10). These individuals commonly respond to corticosteroid therapy (fig1).56 ,59 ,62 ,64-66
Characteristics of the variant syndromes
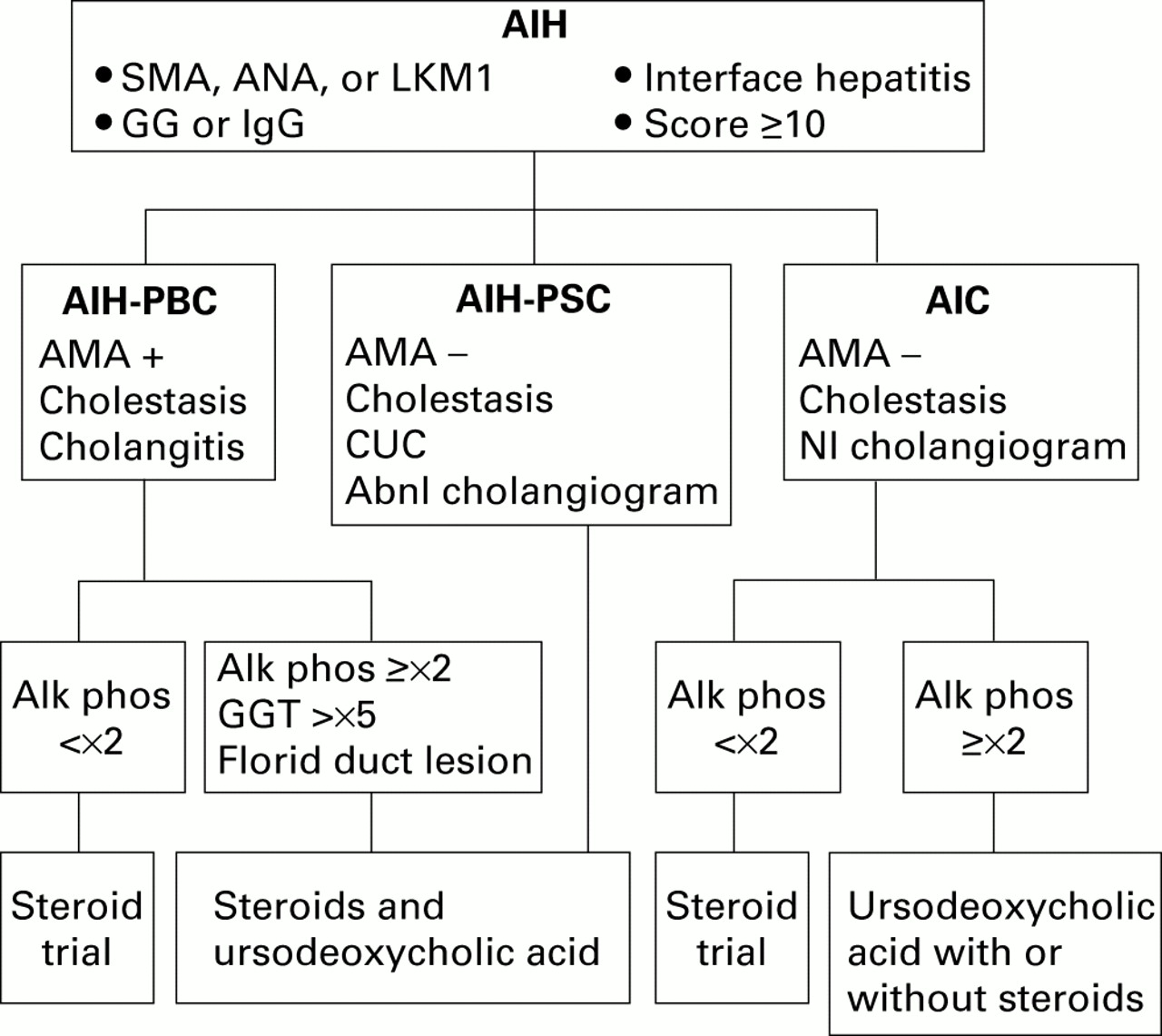
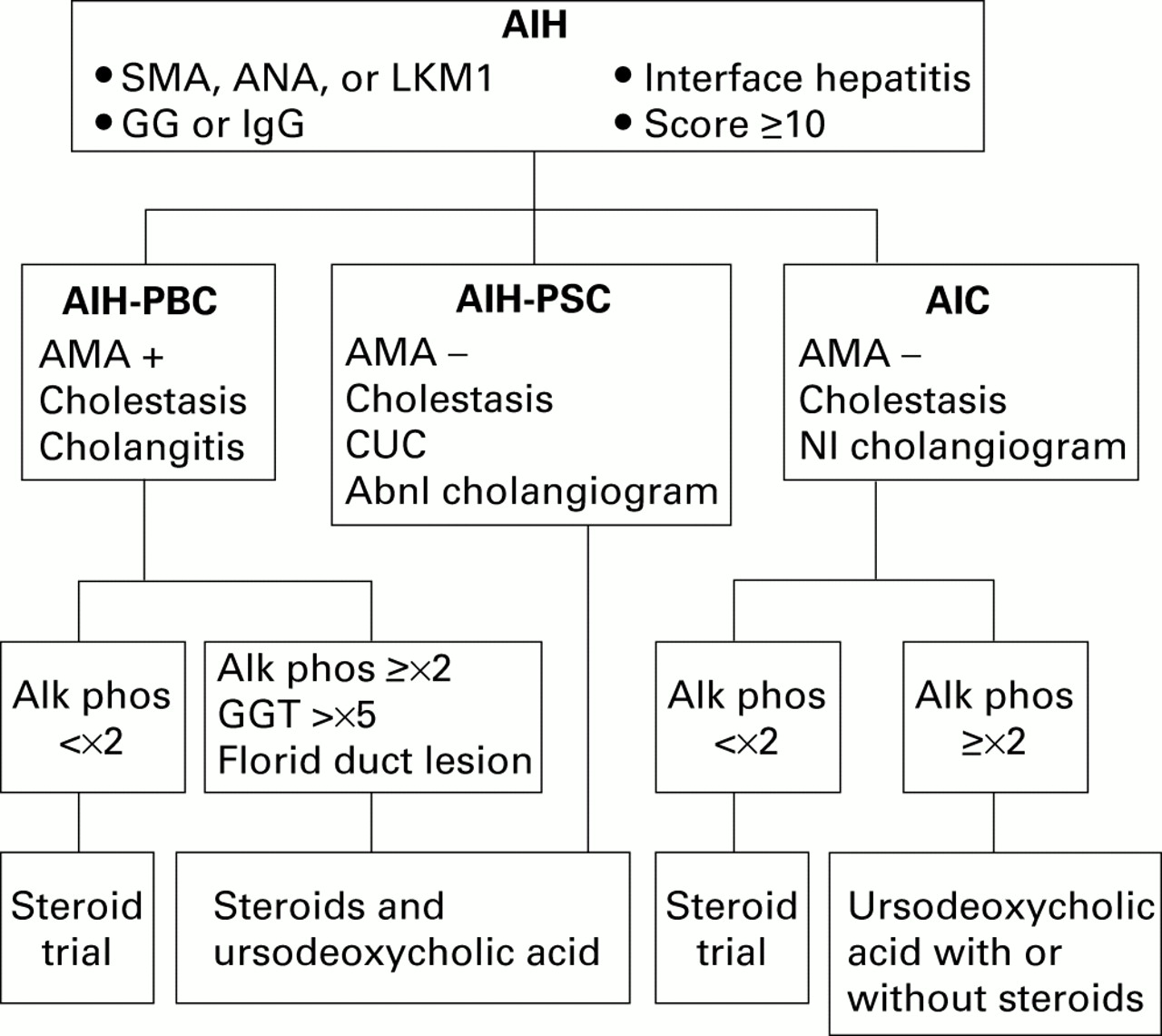{kind=link}
Diagnostic and treatment algorithm for variant syndromes of autoimmune hepatitis (AIH). Seropositivity for smooth muscle antibodies (SMA), antinuclear antibodies (ANA), and/or antibodies to liver/kidney microsome type 1 (anti-LKM1), abnormal serum gamma globulin (GG) or immunoglobulin G (IgG) level, interface hepatitis on histological examination, and/or an aggregate score of at least 10 by the modified system of the International Autoimmune Hepatitis Group support the diagnosis of AIH. Concurrent findings of antimitochondrial antibodies (AMA+), cholestatic clinical and/or laboratory features, and histological evidence of cholangitis indicate an overlap syndrome with primary biliary cirrhosis (AIH-PBC). Cholestatic clinical and/or laboratory features, absence of antimitochondrial antibodies (AMA−), concurrent chronic ulcerative colitis (CUC), and an abnormal (Abnl) cholangiogram indicate an overlap syndrome with primary sclerosing cholangitis (AIH-PSC). Similar clinical, laboratory, and histological findings in the absence of CUC and with a normal (Nl) cholangiogram suggest the outlier syndrome of autoimmune cholangitis (AIC). Treatment decisions are based on the predominant manifestations of the disease, as assessed mainly by serum alkaline phosphatase (Alk phos) or γ-glutamyl transpeptidase (GGT) levels, serum titres of ANA and/or SMA, and histological features.
In contrast, patients with mainly features of PBC have pronounced cholestatic features manifested by serum alkaline phosphatase levels greater than twofold normal, serum γ-glutamyl transpeptidase concentrations at least fivefold normal, and florid bile duct lesions on histological examination (fig 1).67 These individuals commonly achieve a complete biochemical response on a combination of corticosteroids and ursodeoxycholic acid. Cyclosporine A has also been used successfully in one patient who was recalcitrant to both corticosteroids and ursodeoxycholic acid.68
Treatment response may relate to the stage of PBC which is present at accession and the genetic factors that affect outcome in AIH. Early stage PBC may lack the classical histological changes and resemble AIH.69 These patients may yet evolve to classical PBC but at the time of diagnosis they may be responsive to therapy with corticosteroids.65 ,66 ,70 Similarly, the genetic risk factors that affect the susceptibility and outcome of AIH may influence the clinical expression and corticosteroid responsiveness of patients with hybrid features. Individuals with a “hepatitic” form of PBC frequently are positive for HLA B8, DR3, or DR4,66 and these genetic markers can be shared in all forms of autoimmune liver disease with a variable impact on disease behaviour.71-73
A diagnostic algorithm that has been proposed for the AIH-PBC variant requires the presence of at least two of three accepted criteria for each disease in the same patient (fig 1).67 This diagnostic formula is arbitrary, and its sensitivity for the syndrome has not been established. Nevertheless, it does provide a diagnostic template that can be consistently applied. An alternative approach is to designate only those individuals who have scores sufficient for the probable diagnosis of AIH (aggregate scores ⩾10), seropositivity for AMA, and cholangitis on histological examination as AIH-PBC variants (fig 1).56 ,61 This strategy is compromised by revisions of the scoring system for AIH that promise to eliminate incompatible diagnoses.51 More stringent diagnostic criteria for AIH will enlarge the pool of individuals with non-diagnostic findings and increase the heterogeneity of patients with variant syndromes.57 ,58
AIH AND PSC
Histological changes of lymphocytic, pleomorphic, or fibrous cholangitis, concurrent inflammatory bowel disease, and/or failure to respond to corticosteroids are justifications for cholangiography in patients with AIH.56 ,58 ,59 ,61 As many as 41% of these individuals have cholangiographic changes of PSC and are classifiable as AIH-PSC variants (table 1).74 Similarly, 54% of patients with PSC have aggregate scores for AIH that indicate the probability of coexistent AIH and PSC.55 ,56 In 2% of these patients, the diagnostic criteria for definite AIH are satisfied.55 PSC shares genetic risk factors with AIH (HLA B8, DR3, and Drw52),73 ,75-78 and this host related predisposition may result in similar clinical expressions or in transitions between the diseases.79-85 Conversely, HLA DR4, which predisposes to AIH, protects against PSC, and its presence does not support a strong PSC component of the variant syndrome.64 ,73
Diagnostic difficulties occur mainly in children and in adults with normal cholangiograms. “Autoimmune sclerosing cholangitis” is a disorder described in children who have the clinical phenotype of AIH but abnormal cholangiograms.27 ,86-89 Because these children have clinical features of AIH and cholangiographic changes of PSC, they satisfy criteria for an overlap syndrome. Inflammatory bowel disease however is typically absent; cholestasis is neither a biochemical nor a histological feature; and response to corticosteroid therapy is similar to that in classical type 1 AIH.27 ,86 ,88 ,89 Consequently, they are distinct from the AIH-PSC syndrome reported in adults and are best categorised separately until their true nature is fully defined.
Adults with AIH, cholestatic features, seronegativity for AMA, and normal cholangiograms are another diagnostic problem as they may have small duct PSC74 ,90 or autoimmune cholangitis (AIC).64 High serum alkaline phosphatase activity or a ratio of serum alkaline phosphatase level to aspartate aminotransferase level (ALP: AST) that exceeds 1.5 suggests the existence of an AIH-PSC overlap, especially if biliary changes are evident on liver biopsy examination and inflammatory bowel disease is present.83 The scoring system of the International Autoimmune Hepatitis Group has been modified to separate patients with PSC from those of AIH, mainly by assigning no points to ALP:AST ratios of 1.5–3.0 and deducting two points from the aggregate score if the ratio exceeds 3.0.51 Currently, the only distinguishing clinical feature between small duct PSC and AIC is the presence or absence of inflammatory bowel disease.
There is no established treatment for the AIH-PSC variant (fig 1). Corticosteroid therapy has been associated with normalisation of serum aminotransferase levels and improvement in the histological features of inflammation in some patients.81 ,83 In others, corticosteroid therapy has been ineffective.56 ,74Treatment is empiric, and it should be tailored to the predominant manifestations of the hybrid syndrome. The potential for a corticosteroid response is reserved for those individuals with serum alkaline phosphatase levels below twofold normal.56 ,59Ursodeoxycholic acid can be considered for those individuals with dominant cholestatic features but it has generally not been useful in PSC at conventional doses.91 The combination of corticosteroids and ursodeoxycholic acid manages all aspects of the disease and intuitively is the most appealing treatment (fig 1). Its value however is uncertain, and the expectation that two different ineffective therapies will be more successful when administered together is low.
AIH AND VIRAL HEPATITIS
AIH is by definition a non-viral disease50 ,51 but there is an association between viral infection and the autoimmune response.92 Autoantibodies commonly occur in chronic hepatitis B and C infection.93-101 Indeed, ANA and/or SMA occur in 20–40% of patients with chronic hepatitis B or C97 ,99; anti-LKM1 are detectable in up to 6% of adult patients101-108 and 10% of children with chronic hepatitis C22 ,98; antibodies to LKM3 are found in 13% of patients with chronic hepatitis D107 ,109; immune diseases that are viral-antigen driven (cryoglobulinaemia) or autoantigen driven (autoimmune thyroiditis, Sjogren's syndrome) frequently coexist110-116; and corticosteroids may be effective in some patients.117-123 In most individuals, the viral components predominate, and these patients are best designated as “chronic viral hepatitis with autoimmune features” and treated accordingly.124 Rarely, the autoimmune features predominate, and the concurrent true viral infection is coincidental or aetiological but non-essential for perpetuation of the disorder.118 ,119 ,125 These latter patients constitute the overlap syndrome of AIH and true viral infection (table1).57 ,58 ,126 Their diagnosis requires the clinical, laboratory, and histological features of classical AIH.
Candidates for designation as an AIH-viral hepatitis overlap are patients with high titre SMA and/or ANA (titres, ⩾1:320) and hypergammaglobulinaemia (table 1).125 ,126 These patients commonly are HLA DR3 positive, and examination of the liver tissue usually discloses moderate to severe interface hepatitis with or without panacinar hepatitis.125 ,127 ,128 Titres of anti-LKM1 do not distinguish patients with and without chronic hepatitis C,22 ,123 and anti-LKM1 in any titre may denote an AIH-viral hepatitis overlap syndrome. Histological features of viral infection, such as steatosis and portal lymphoid aggregates, are typically absent in patients with the AIH-viral hepatitis variant (fig1).125 ,129 ,130
Multiple studies have now indicated that the rare patients with AIH-viral hepatitis overlap can respond to corticosteroid treatment (fig 1).118 ,120-123 Anecdotal reports of exacerbations of inflammatory activity or worsening of autoimmune manifestations after interferon treatment have justified this alternative management strategy in highly selected patients.123 ,131-136Instances of deterioration during interferon treatment however have been rare, and the routine precautionary use of corticosteroids in individuals without definite AIH is unjustified.96 ,136-141 These patients however must be monitored closely during interferon therapy, especially those with anti-LKM1, for exacerbation of their disease.101Fortunately, the AIH-viral hepatitis variant is rare and it may be a serendipitous clustering of separate diseases rather than a single entity.
AUTOIMMUNE CHOLANGITIS
Autoimmune cholangitis is an “outlier” rather than an “overlap” syndrome.56-59 Clinical and/or laboratory features of cholestasis are present as are high titres of ANA and/or SMA. Antimitochondrial antibodies are undetectable, and bile duct injury is evident in liver tissue. The diagnosis implies the absence of inflammatory bowel disease and/or a normal cholangiogram (table1).56-59
Retrospective analyses that have used patients with PBC as discovery fields for the diagnosis have emphasised its resemblance to PBC. In these studies, patients with AIC have been indistinguishable from those with PBC by histological examination142; display of pyruvate dehydrogenase complexes on biliary epithelia143; presence of PBC specific antibodies to 2-oxo-acid dehydrogenase complex in blood144-146; clinical and laboratory features147; and responsiveness to ursodeoxycholic acid.148 Conversely, retrospective analyses using patients with AIH as discovery fields for the diagnosis have emphasised its resemblance to AIH. In these studies, patients with autoimmune cholangitis have had ANA and/or SMA,149-151 interface hepatitis and portal plasma cell infiltration,149 ,151high mean diagnostic scores for AIH,56 ,58 ,64 ,152 and occasional improvement after corticosteroid therapy.151
Autoimmune cholangitis is probably a heterogeneous syndrome that includes patients with AMA negative PBC, small duct PSC, AIH with bile duct damage, concurrent AIH and small duct PSC, and various transition states.64 Prospective studies have emphasised the inability to classify these individuals into a single diagnostic category. Histological findings may be indistinguishable from PBC or PSC, and individual patients cannot be discriminated by clinical, laboratory, genetic, or histological parameters.64Similarly, responsiveness to corticosteroids or ursodeoxycholic acid is variable and generally poor. Indeed, most studies emphasise an inability to induce histological improvement with either drug.64 ,150 ,151 Treatment is empiric and consists of corticosteroids, ursodeoxycholic acid, or a combination of both (fig1). Therapy should be reserved mainly for those individuals who are symptomatic with jaundice, pruritus, and/or malaise.
Summary
The characteristic features of the variant syndromes of AIH are summarised in table 1. The presence of AMA, histological features of cholangitis, abnormal cholangiogram, disproportionate elevation of the serum alkaline phosphatase level, laboratory features of cholestasis, and/or concurrent true viral infection in individuals who otherwise have classical features of AIH constitute the variant forms.
The variant syndromes are in part the result of improved diagnostic criteria that exclude patients with cholestatic and viral features from the diagnosis of AIH. Their frequency emphasises the variability of the classical syndromes of autoimmune liver disease, and it suggests that the classical disorders share genetic predispositions and/or pathogenic mechanisms that can result in hybrid syndromes, concurrent diseases, or transition states. The variant syndromes should be classified separately until their nature is fully defined. In this fashion, the natural history of the established disorders can be preserved, and the pathogenic pathways of the newly recognised entities examined. Treatments are empiric until diagnostic criteria can be codified and multicentre clinical trials instituted. The variant syndromes may reflect the “larval stage” of a classical syndrome, a collage of different autoimmune manifestations in a susceptible host, a unique expression of coincidental factors that are unrelated, or an independent complex disorder with a heterogeneous phenotype. Each syndrome should stand alone until its true identity is revealed.
Abbreviations used in this paper
- AIH
- autoimmune hepatitis
- AIC
- autoimmune cholangitis
- ANA
- antinuclear antibodies
- SMA
- smooth muscle antibodies
- HLA
- human leucocyte antigen
- anti-LKM1
- antibodies to liver/kidney microsome type 1
- APS1
- autoimmune polyglandular syndrome type 1
- anti-SLA
- antibodies to soluble liver antigen
- anti-LP
- antibodies to liver pancreas
- anti-SLA/LP
- antibodies to soluble liver antigen/liver pancreas
- tRNP(Ser)Sec
- transfer RNA-protein complex
- PBC
- primary biliary cirrhosis
- PSC
- primary sclerosing cholangitis
- AMA
- antimitochondrial antibodies
- ALP:AST
- serum alkaline phosphatase level to serum aspartate aminotransferase level