Article Text

Statistics from Altmetric.com
- ECM, extracellular matrix
- EGF, epidermal growth factor
- FAK, focal adhesion kinase
- HGF, hepatocyte growth factor
- IBD, inflammatory bowel disease
- IFN, interferon
- KGF, keratinocyte growth factor
- MMP, matrix metalloproteinase
- NADPH, nicotinamide adenine dinucleotide phosphate hydrolase
- PDGF, platelet-derived growth factor
- SMA, smooth-muscle actin
- SOCS, suppressor of cytokine signalling
- TGFβ, transforming growth factor β
- TIMP, tissue inhibitor of metalloproteinase
- TNF, tumour necrosis factor
The mechanisms of wound healing in general have gained interest in recent years, as it has become obvious that the tightly regulated process of tissue repair and regeneration is of great importance for organ homeostasis. Insufficient as well as excessive tissue repair both impair gastrointestinal function. Formation of ulcers and fistulas on the one hand, and of fibrosis and stricture on the other, represent just two sides of one medal.
So far, the physiological pathways involved in intestinal wound healing are only partially understood. During acute and chronic intestinal inflammation, macrophages and neutrophils induce local tissue damage by secreting reactive oxygen radicals and tissue-degrading enzymes. This is followed by the release of pro-inflammatory cytokines, as well as chemotactic and cell-activating peptides previously bound to the matrix. If tissue damage is severe, myofibroblasts migrate to the sites of the defect. This migratory function, the ability to contract the wound area and the production of extracellular matrix (ECM) by intestinal myofibroblast cells certainly have important roles in the physiological situation and are altered by chronic inflammation. Available treatments of intestinal strictures, fibrosis and fistulas are insufficient and unsatisfactory. New therapeutic approaches are urgently needed. Future intervention should involve stronger and more selective prevention of the continuous tissue damage and a change in wound healing by modulation of myofibroblast migration and ECM synthesis.
Severe mucosal tissue damage requiring efficient wound healing is a main feature of inflammatory bowel disease (IBD), with its two entities Crohn’s disease and ulcerative colitis. During radiation enteritis1,2 or chronic ischaemic enteritis,3,4 the bowel wall is similarly damaged, with subsequent inflammation. Furthermore, cystic fibrosis also may lead to colonic wall thickening, fibrotic colonopathy and stricture formation.5,6,7,8,9,10 In rare cases, recurrent diverticulitis also may cause colonic strictures.11,12 During collagenous colitis, fibrosis typically occurs at the basement membrane directly beneath the epithelial barrier.13–15
Despite an extensive and detailed investigation on the immunological pathways involved in chronic inflammation in recent years, the physiology and pathophysiology of mucosal wound healing has remained widely unexplored. This is surprising because insufficient (abscess, fistula) or excessive wound healing (fibrosis) is the main indication for surgery in patients with Crohn’s disease.16–21 The therapeutic problems caused by bowel wall fibrosis are common18: about 75% of all patients with Crohn’s disease have to undergo surgery at least once during the course of their disease.22,23 In half of these patients, intestinal obstructions and strictures are the indications for surgery. This means that intestinal obstructions require abdominal surgery in about one third of all patients with Crohn’s disease.22,23 In >45% of patients with Crohn’s disease, these obstructions are recurrent.24 Owing to limited pathophysiological insights, good conservative treatment options are unavailable to prevent stricture formation and fibrosis. In contrast with anti-inflammatory treatment, little therapeutic progress has been made with respect to intestinal fibrosis.25 Current preventive attempts therefore rest primarily on long-term anti-inflammatory treatment.16,26,27 However, this mainly anti-inflammatory approach is often ineffective, leading to surgery and stricturoplasty, which remain the major treatment methods for intestinal fibrosis.28 Unfortunately, even the surgical approach is often only associated with short-term resolution of symptoms, as strictures tend to recur. Several non-surgical procedures for the treatment of strictures, such as balloon dilatation17,29–35 or polyvinyl over-the-guidewire dilatation,36 have been reported, which are still controversial. Injection of glucocorticoids into the strictures after dilatation also has been suggested.37 An effective prevention of fibrosis and stricture formation, therefore, would be a major progress.
At this point in time, we are just beginning to understand the mechanisms that lead from intestinal inflammation to fibrosis. Current concepts view fibrosis as a reactive process. A chronic or recurrent inflammation is considered a necessary precondition for the initiation of intestinal fibrosis. In this view, fibrosis is a pathologically augmented healing response to inflammation-induced destruction and injury of mucosal tissue. However, what triggers increased fibrosis in some patients and not in others is still unclear.
As mesenchymal cells—fibroblasts, myofibroblasts and smooth-muscle cells—are the main producers of ECM components that are deposited during fibrosis, these cells may be regarded as one central player or the effectors of intestinal fibrosis. Therefore, after highlighting the mechanisms involved in inflammatory tissue damage, the role of fibroblasts and myofibroblasts during intestinal fibrosis will be discussed. In the authors’ view, there is no primary defect or malfunction in these cells that leads to fibrosis. They react to the given intestinal environment with enormous plasticity, but exactly in the way they are programmed to. Therefore, we have to understand these conditions and mediators, as well as the primary programming of intestinal mesenchymal cells, to be finally able to develop new treatment approaches.
HOW IT STARTS: TISSUE DAMAGE CAUSED BY INTESTINAL INFLAMMATION
Inflammation is associated with an infiltrate of immune cells, such as T cells, macrophages and neutrophils, and it also often causes severe damage to the tissue in which it occurs. In the case of intestinal mucosa, severe inflammation is followed by a loss of epithelial cells and a degradation of ECM in the lamina propria, clinically leading to ulcerations (fig 1). Enzymes and mediators mainly secreted by monocytes, intestinal macrophages and granulocytes are responsible for this tissue damage. This continuous inflammation and tissue degradation may consequently lead to fibrosis and stricture formation (fig 1).

Fibrosis in the colon: the clinical problem. Ulcerations and tissue damage (black arrow) are caused by chronic inflammation. This is followed by bowel wall fibrosis, leading to pseudopolyps (blue arrow) or strictures reducing the colon lumen (white arrows).
Oxidants are important contributors to mucosal, and eventually submucosal, tissue destruction (fig 2). Oxygen metabolites, such as oxygen or hydroxide radicals, are produced in large amounts by infiltrating leucocytes in the inflamed mucosa.38 The normal intestinal wall contains relatively small amounts of antioxidative enzymes.39 The production of superoxide and other reactive oxygen intermediates is catalysed by a membrane-associated nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.40 The enzyme system responsible for superoxide generation forms a small transmembrane electron transport system that results in the oxidation of NADPH on the cytoplasmic surface and the generation of a superoxide on the outer surface of the membrane. It is a heterodimer composed of a 91-kDa glycoprotein (termed gp91-phox, for the 91-kDa glycoprotein of phagocyte oxidase) and a 22-kDa polypeptide (p22-phox).41 In addition to the cytochrome b heterodimer, four cytosolic factors (p47-phox, p67-phox, p40-phox and p21 Rac) are required for NADPH oxidase activity. The enzyme is dormant in resting cells and becomes active on stimulation. Induction of NADPH oxidase expression in intestinal macrophages from inflamed mucosa associated with increased oxidative burst activity has recently been shown.42 Inhibition of this oxygen radical secretion ameliorates experimental colitis in mice (unpublished data).
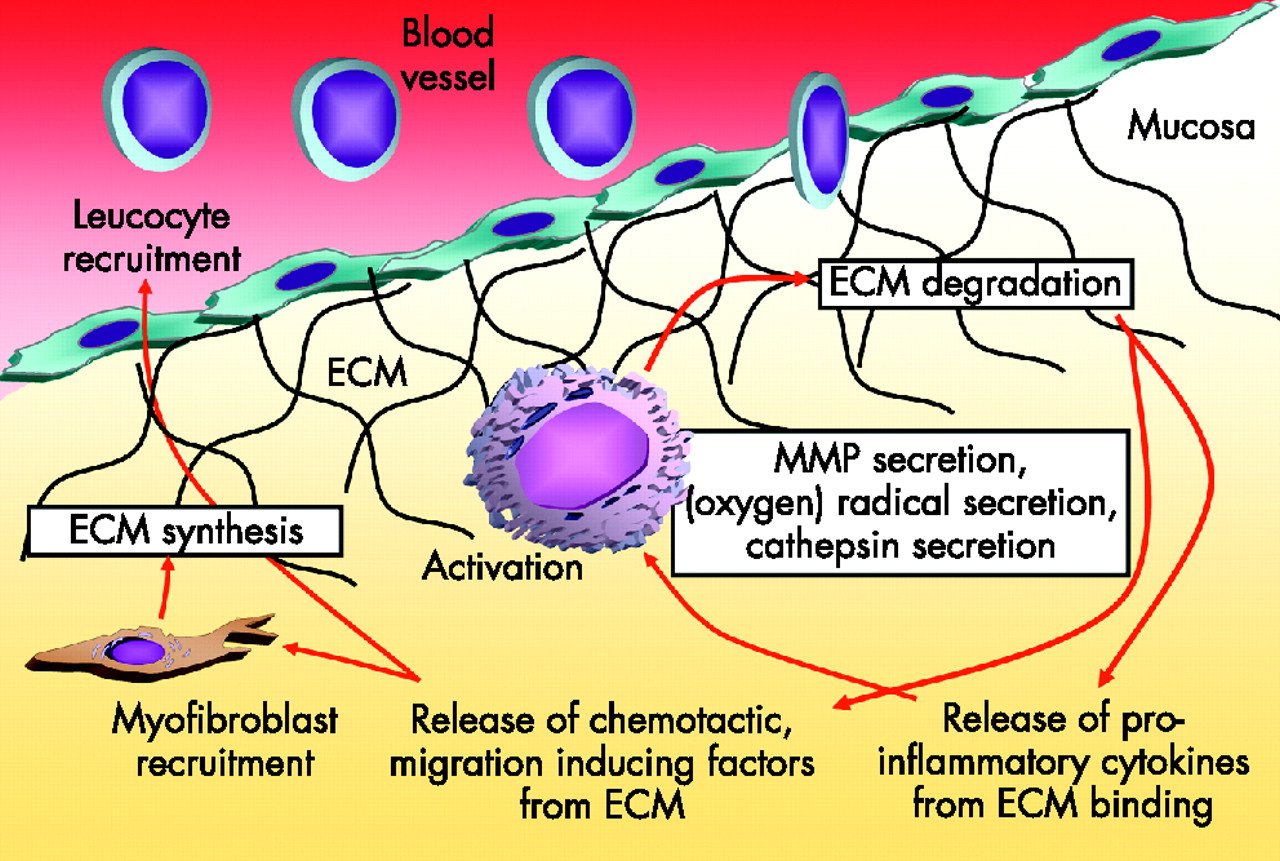
Inflammation and degradation of extracellular matrix (ECM). During severe and chronic intestinal inflammation, activated local macrophages or neutrophils secrete oxygen radicals, cathepsins and matrix metalloproteinases (MMPs), leading to degradation of ECM. This is followed by a release of pro-inflammatory cytokines from ECM-binding sites, which further activate the leucocytes. In addition, chemotactic, migration-inducing factors are released from ECM, causing an increase in infiltration of leucocytes evading from the blood vessel through the endothelium, finding a paved way through the degraded ECM. The same factors attract myofibroblasts, which synthesise ECM, in an attempt to limit the process.
Besides radical formation, infiltrating and locally activated immune cells respond to intestinal inflammation by secreting ECM-modifying and ECM-degrading enzymes43 (fig 2). This permits further infiltration of immune and non-immune cells into the inflamed area, finally paving the way for the migration of myofibroblasts (fig 2). Two major groups of proteinases are secreted: serine proteases such as elastases or collagenases (degrade elastin, fibronectin, laminin, collagen and proteoglycans) and matrix metalloproteinases (MMPs). In contrast with MMPs, which are usually secreted as inactive pro-forms (zymogens), elastases are stored intracellularly in neutrophils and monocytes or in macrophages in their active form.43 The MMP family of enzymes consists of at least 15 distinct members, nine of which are expressed in leucocytes. All MMPs degrade ECM components. Their activity is regulated by inhibiting enzymes called tissue inhibitors of metalloproteinases (TIMPs). The balance between the tissue-degrading MMPs and their inhibitors is important for the degree of intestinal wall damage during inflammation.
By analysing differences in gene expression between intestinal macrophages from normal and inflamed mucosa with microarrays and subtractive hybridisation, a dramatic induction of tissue-degrading enzymes can be detected. The expression levels of NADPH oxidase42 and also of cathepsin D44 were largely increased (fig 2). The proteolytic and destructive properties of the cathepsins have a role in several chronic inflammatory diseases. Cathepsin family members are involved in the remodelling of ECM proteins under inflammatory conditions.45 Cathepsins L and E degrade collagen and elastin, and have a role in the pathogenesis of artheriosclerosis and lung emphysema/fibrosis.45 Destruction of elastin-rich tissue is furthermore associated with local accumulation of macrophages containing high levels of cathepsins B and L. The aspartic proteinase cathepsin D also has the potential to initiate a proteolytic cascade, and to degrade and remodel ECM. Mononuclear cells expressing cathepsins B and L were shown to have important roles in patients with rheumatoid arthritis, where they take part in joint destruction and bone erosion. An important part of the cathepsin family members B, D and L during intestinal inflammation and tissue damage is evident, as their inhibition in dextran sodium sulphate-induced colitis is followed by a considerable amelioration of the disease.46
Under physiological conditions, the process of tissue damage caused by acute inflammation comes to an end, and is followed by wound healing to a restitutio ad integrum. If the basement membrane underlying the injured epithelium is intact, residual epithelial cells at the edge of the wound become motile. They move along the basement membrane until they meet adjacent epithelial cells to form new tight junctions. This process is called “restitution”.47,48 Restitution is supported by growth factors secreted by mesenchymal (as well as epithelial) cells, such as hepatocyte growth factor, keratinocyte growth factor, epidermal growth factor, transforming growth factor β (TGFβ), and acidic and basic fibroblast growth factors.49–52
GENETIC CONTROL OF WOUND HEALING AND FIBROSIS?
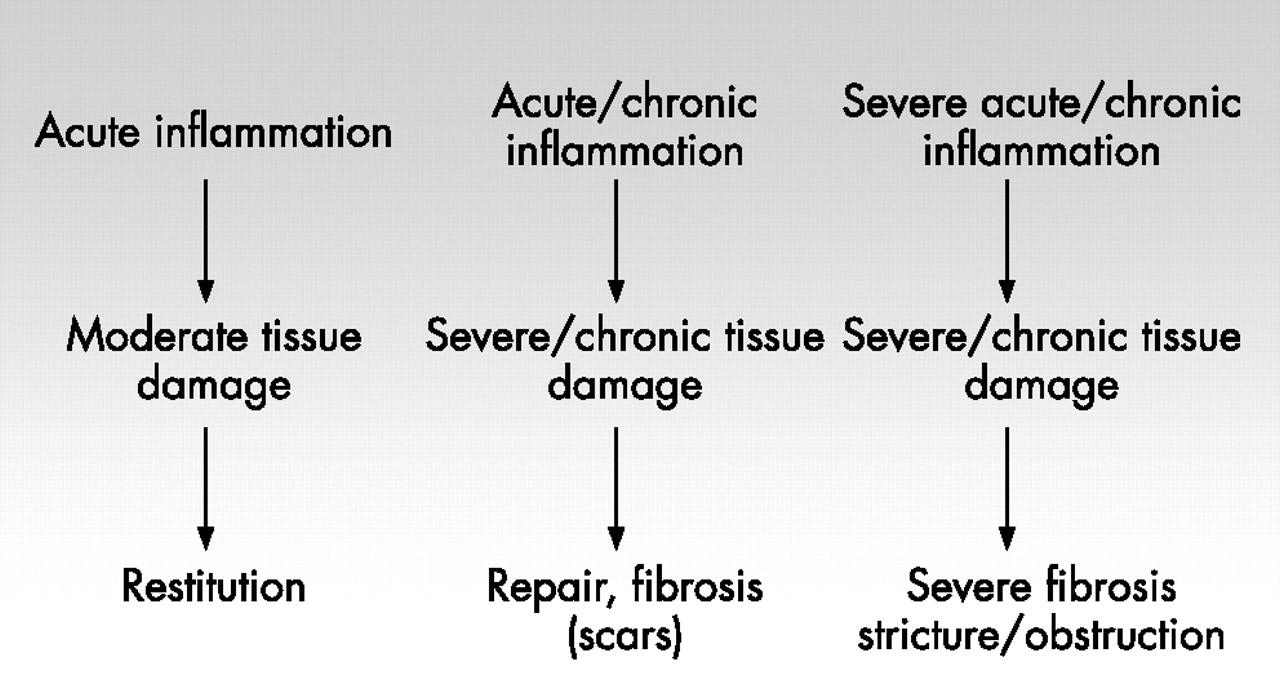
If the defect is deeper, with subepithelial tissue damage, the area below the basement membrane has to be reconstituted in addition to the epithelial surface (fig 3). One of the key events in that process is the contraction of the underlying lamina propria to limit the wound area the epithelium finally has to cover. A rapid wound closure is important to reduce the time of impaired barrier function of the intestinal wall. Recent studies have provided evidence for the deleterious consequences of an uncontrolled and longlasting translocation of bacteria from the gut lumen into the mucosal wall.53 It is crucial to prevent bacterial translocation, or—if impossible—to rapidly detect and sense translocated bacteria, a lesson we learnt from the first susceptibility gene for Crohn’s disease, NOD2/CARD15.53 In fact, variants of NOD2/CARD15 causing an increased risk of developing Crohn’s disease are also associated with a higher frequency of fibrosing and stricturing disease.54–59 Further clinical evidence suggests a genetically determinated risk to develop strictures. Some patients obviously have rapidly recurring strictures, whereas others permanently have an inflammatory, non-stricturing disease type (fig 4).

Severity of inflammation and tissue repair. Acute intestinal inflammation is normally followed by moderate or limited tissue damage and complete restitution. A more severe acute or moderate chronic inflammation may result in severe or chronic tissue degradation and damage, followed by repair, and may also be accompanied by fibrosis and scars. However, severe acute and longlasting chronic tissue damage may be associated with severe fibrosis, leading to intestinal strictures and obstruction.

Stricture formation. Genetic influence or consequence of aggressive inflammation?
Messenger RNAs encoding for toll-like receptors 1–9, as well as for NOD1 and NOD2, could be amplified from cultured primary human intestinal myofibroblasts. After stimulation with bacterial wall products, an up to fourfold up regulation of toll-like receptors and elements of the signal transduction cascade (MyD88, Toll/interleukin 1 receptor domain-containing adapter protein) were observed. Therefore, intestinal myofibroblasts themselves can respond to bacterial products that have penetrated into the subepithelial compartment.60
Is the process of wound healing and stricture formation directly influenced by bacterial translocation and the expression of so-called pattern recognition receptors responsible for the rapid detection of translocated bacteria? We do not know at present. There are different explanations for how impaired recognition of bacterial translocation could cause more severe inflammation and stricture formation. The presence of bacterial wall components might cause a secondary excessive up regulation of pro-inflammatory transcription factors, such as nuclear factor κB, which is mainly activated in macrophages and epithelial cells in the chronically inflamed mucosa.61 This might be followed by prolonged macrophage activation and induction of NADPH oxidase expression,42 leading to increased oxygen radical secretion to destroy the invaded bacteria. The higher concentration of radicals would cause additional tissue destruction and an extension of the wound areas bringing the “reconstitution system” to its limits. Translocated but not efficiently removed bacteria, on the other hand, could directly stimulate neighbouring mesenchymal cells via pattern recognition receptors leading to increased activation.62
MYOFIBROBLASTS IN THE INTESTINAL MUCOSA
After injury, tissue repair and wound healing take place, involving new tissue formation and scar constitution.63 Owing to their contractile and migratory ability and their competence in secreting matrix proteins and growth factors, fibroblasts, myofibroblasts and smooth-muscle cells have an important role in both the reconstitution and the repair processes.
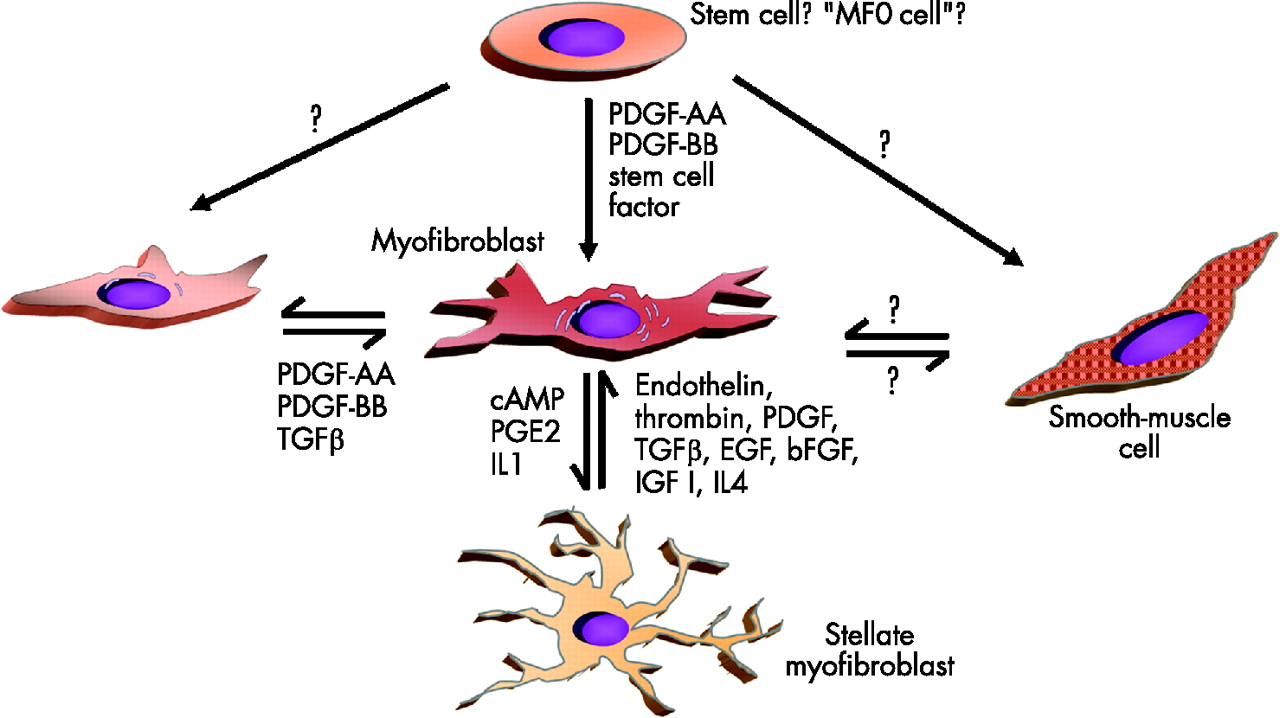
Recent years have generated evidence that there is a phenotypic heterogeneity among fibroblasts in general and specifically among intestinal wall fibroblasts.48,64–66 Some fibroblast types have features of smooth-muscle cell differentiation. These cells were termed myofibroblasts.67 Myofibroblasts play an important part in tissue growth and development in different tissues.13,68 They are central players during tissue repair that may finally lead to local or generalised fibrosis and stenosis (fig 5).

Hypothetical transdifferentiation pathways of myofibroblasts. It has been speculated that subepithelial myofibroblasts and interstitial cells of Cajal-like T cells differentiate from a common precursor (an “MF0 cell”). Whereas myofibroblasts clearly originate from fibroblasts, it has not been proved that they have the ability to differentiate into or transdifferentiate from smooth-muscle cells. In the liver, activated myofibroblasts can transdifferentiate into stellate cells. bFGF, basic fibroblast growth factor; EGF, epidermal growth factor; IGF, insulin-like growth factor; IL, interleukin; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2; TGF, transforming growth factor. (Modified according to Powell et al.71)
Whether myofibroblasts are a distinct cell type or a differentiation state of fibroblasts is still debatable.69 They are defined morphologically and immunologically mainly by the expression of cytoskeletal proteins.13 Among these specific features are prominent cytoplasmic actin microfilaments (stress fibres) and the formation of intercellular connections via adherens and gap junctions.70 The simplest definition used in the literature is based on the expression of α-smooth-muscle actin (α-SMA). Some authors see myofibroblasts as an intermediate state between fibroblasts and smooth-muscle cells (fig 5). However, it is unknown whether they originate from one of these two cell types in vivo. Other classifications use filament proteins as a means of differentiation also to define myofibroblasts. Besides α-SMA, vimentin and desmin are often described.71 On the basis of the expression of filament protein, Powell71 proposed a classification of myofibroblasts: myofibroblasts expressing only vimentin are “V type”, those expressing vimentin and desmin are “VD type”, those additionally expressing α-SMA are “VAD type”, those expressing vimentin and α-SMA are “VA type” and those expressing vimentin and myosin are “VM type”. Whether these phenotypes and differences in the expression of cytoskeletal proteins are associated with functional differences is presently unknown.
In the intestinal mucosa, two types of myofibroblasts are found under physiological conditions. Besides the subepithelial myofibroblasts, the interstitial cells of Cajal have a myofibroblast phenotype (fig 6). The interstitial cells of Cajal are located in the submucosa and muscularis propria in association with the smooth-muscle layer of the gut. It is unknown whether subepithelial myofibroblasts and interstitial cells of Cajal-like T cells differentiate from a common precursor (an MF0 cell, or which factors induce this differentiation fig 5). Both types of myofibroblasts form a network or “syncytium”; however, it is unclear whether or not both networks are connected. The subepithelial myofibroblasts mainly located at the bases of intestinal crypts in the lamina propria are morphologically well characterised. Their functional capacity is nevertheless widely unexplored.71 They form a three-dimensional network and communicate with each other by gap and adherens junctions, but also keep up connections with epithelial cells through fenestrations in the basement membrane. They also interact with intestinal macrophages. Whereas the interstitial cells of Cajal have the V or VM phenotype, the subepithelial myofibroblasts have the VA type71 (fig 6). Platelet-derived growth factor (PDGF) seems to be essential for the differentiation and development of myofibroblasts (fig 5). Besides PDGF and TGFβ, insulin-like growth factor I (IGFI) and interleukin (IL)4 may be factors relevant for the transdifferentiation of fibroblasts to myofibroblasts72 (fig 5).

Characteristics of the two types of myofibroblasts found in the bowel wall. IEC, intestinal epithelial cells; SMA, smooth-muscle cell actin; SMC, smooth-muscle cell.
ARE MYOFIBROBLASTS THE ONES TO BLAME?
The intestinal subepithelial myofibroblasts have ultrastructural characteristics of activated cells. In other tissues such as the liver, the differentiation or activation of fibroblasts into myofibroblasts is an early event in the development of fibrosis.63 Their prolonged presence and over-representation are hallmarks in the pathophysiology of tissue fibrosis in several organs.73 Myofibroblasts can influence epithelial cell proliferation and differentiation. This may become important during wound healing or in general after mucosal damage in mucosal inflammation.74,75
Despite the characteristics of “activated” cells, it must be kept in mind that subepithelial myofibroblasts display these characteristics physiologically and that usually an activation means a change of function from a resting or inactive state to increased secretion of cellular products. This change does not occur for subepithelial myofibroblasts and it is somewhat a contradiction to talk about constitutively activated cells.
In the liver, several conditions or pathways lead to an activation of fibroblasts or stellate cells into myofibroblasts. Usually, this activation is determined by immunostaining for α-SMA. Presence of α-SMA staining in fibroblast-like cells is thought to be correlated with cell activation. Among the conditions, which are followed by α-SMA induction, is the simple culture of fibroblasts in serum-containing growth media. Whereas TGFβ seems to be the most potent and important inductor of α-SMA, other cytokines such as IL1, tumour necrosis factor (TNF), PDGF and basic fibroblast growth factor can also induce α-SMA expression.16,60,76–80
IGFI has been reported to stimulate smooth-muscle cell hyperplasia.81 IGFI and TGFβ are both up regulated in myofibroblasts at sites of fibrosis in experimental enterocolitis and in patients with Crohn’s disease.82 Both growth factors can induce type I collagen expression; however, only TGFβ potently stimulates α-SMA expression. On the other hand, IGFI may stimulate the proliferation of TGFβ-activated myofibroblasts without reversing the activated fibrogenic phenotype.82
An important source of TGFβ is epithelial cells hallmarking another cell–cell interaction in intestinal inflammation and fibrosis.78 The rapid activation of fibroblasts or myofibroblasts requires, or is supported by, the presence of ECM molecules. The degradation of fibronectin by MMPs secreted by macrophages (fig 2), in particular, may allow the interaction of fibroblasts or myofibroblasts with the extra domain (ED)-A domain of fibronectin, followed by a rapid stimulation of the mesenchymal cells.83 This ED-A domain of fibronectin occurring during tissue damage may even be necessary for the TGFβ-triggered expression of α-SMA. The splicing variant of fibronectin, ED-A, is an important inducer of migration. Interestingly, the analysis of fibronectin protein in the intestinal mucosa by immunohistochemistry clearly showed reduced amounts of splicing forms of fibronectin, ED-A and ED-B, in inflamed mucosa of patients with Crohn’s disease. In fistulas, the splicing forms ED-A and ED-B were virtually absent. In fibrotic mucosa, the protein of the isoforms was increased. These results were supported by real-time polymerase chain reaction.84
A fibrogenic myofibroblast type is generally believed to exist when intestinal fibrosis is discussed.25 Plenty of clinical and experimental evidence suggest that during normal wound healing, fibroblasts may acquire the morphological and biochemical features of smooth-muscle cells, including expression of α-SMA.25
MYOFIBROBLAST MIGRATION
Migration of myofibroblasts into and through the ECM seems to be a fundamental event during the initial phase of wound healing. After the wound has been repopulated and the chemotactic gradient caused by mediators secreted by inflammatory cells has decreased, fibroblast migration will normally cease. Subepithelial myofibroblasts can stimulate their own migration by autocrine and paracrine factors.85,86 PDGFA, PDGFB, TGFβ1, IGFI and epidermal growth factor can enhance intestinal myofibroblast migration.85 All of these factors cause a dose-dependent increase in intestinal myofibroblast migration. In addition to a chemokinetic effect, growth factors obviously can also stimulate a chemotactic movement.85 These growth factors alone, however, cannot induce intestinal myofibroblast migration. Fibronectin (that can be synthesised by myofibroblasts in large quantities) is essential and mainly responsible for the autocrine induction of migration of intestinal myofibroblats.86 Increasing concentrations of fibronectin induce the migration of intestinal myofibroblasts in a dose-dependent manner. Fibronectin is essentially required for the induction of migration of these cells.86
The migratory response of subepithelial myofibroblasts isolated from the mucosa of patients with Crohn’s disease is markedly reduced when compared with that of cells isolated from controls.87 In myofibroblasts from normal mucosa, a similar persistent reduction in migration can be induced by incubation with TNF or interferon (IFN)γ.87 How this reduced migratory potential contributes to fibrosis in inflammatory bowel disease is still unclear. In other organs, fibroblasts isolated from diseased tissue often have either an enhanced or a reduced migratory potential, depending on the type of disease. Suganuma et al88 examined fibroblasts from patients with interstitial lung fibrosis associated with a collagen vascular disease and from patients with sarcoidosis. Fibroblasts from tissues with dense fibrosis showed a higher migratory potential towards PDGF than that of fibroblasts from a tissue in an earlier stage of fibrosis.88 Pontz et al89 screened fibroblast strains derived from patients with mucopolysaccharidosis. Whereas the synthesis of major ECM components was close to normal, the response of the fibroblasts from patients with mucopolysaccharidosis to chemotactic stimuli was greatly reduced. These studies indicate that changes in the migratory potential of mesenchymal cells are often associated with the development of fibrosis in different organs. However, they also show that the specific pathogenesis of each disease can either increase or reduce this migratory potential. It may be speculated that fibroblast and myofibroblasts respond in a programmed way to the conditions they find in the tissue. They therefore are not to blame for the initiation of tissue fibrosis. They react as they are programmed, so the reasons for the ongoing activation of myofibroblasts need to be elucidated.
Which locally produced mediators are known to modulate myofibroblast migration? TGFβ1 treatment of intestinal myofibroblasts enhances α-SMA expression in these cells, but decreases the migratory potential (our unpublished data). A migration inhibitory effect of IFNγ on normal embryonic and adult mesenchymal cells has been reported.90,91 This could be one explanation for the reduced migration of myofibroblasts isolated from chronically inflamed mucosa, as they are exposed to high levels of IFN for a long time. The induction of migration is a receptor-mediated process, and the interaction between fibronectin and its receptors is the crucial factor.85,86 As IFN affects the cellular distribution of fibronectin, inducing a more extensive fibronectin filament network, IFN may affect the migration of fibroblasts via its effect on fibronectin organisation.87 IFN also affects the cytoskeleton, which is a fundamental organ of cell locomotion.92
TNF can function as a chemoattractant and an activator of migration for skin fibroblasts.93 However, intestinal myofibroblasts respond differently to TNF and show reduced migration under the influence of TNF.87 Thus, the influence of TNF on myofibroblast migration seems to be tissue specific.
Tyrosine phosphorylation of p125 focal adhesion kinase (FAK) is a central regulator of cell migration in health and disease (fig 7).94 FAK is a non-receptor protein tyrosine kinase involved in integrin-mediated control of cell behaviour. The amounts of both cytosolic FAK protein and its phosphorylation normally increase during cell migration.95 FAK becomes phosphorylated at multiple sites, including tyrosine 397 (fig 7), which is a marker for the activation of FAK. Phosphorylation at this residue creates an Src homology 2-binding site for different signalling and adapter proteins.95 FAK-deficient fibroblasts exhibit defects in cell migration and a raised number of cell–substratum contact sites.

Mechanisms of myofibroblast migration. Integrin receptors extracellularly mediate contacts to the extracellular matrix (ECM) and also provide ECM signals for the induction of migration. Focal adhesion kinase (FAK) is associated with integrins, and after activation is phosphorylated, consecutively leading to activation of several kinases in a subsequent signal transduction cascade. As a consequence, the actin cytoskeleton is remodelled and a focal contact point containing high numbers of integrin molecules is moved in the membrane, thereby allowing cell migration.
The persistent functional change of intestinal myofibroblasts isolated from inflamed mucosa of patients with Crohn’s disease or ulcerative colitis, reflected by a reduced migratory potential, is accompanied by a reduction in FAK protein and FAK phosphorylation. This is also persistently inducible in myofibroblasts from control mucosa by pre-exposure to TNF and IFNγ.87 These data suggest that the migration of mucosal myofibroblasts is controlled by the regulation of FAK protein and FAK phosphorylation, and decreased FAK and FAK autophosphorylation may be responsible for a reduced migratory capacity of IBD-myofibroblasts and TNF/IFNγ-treated cells. FAK protein is regulated post-translationally in these cells because no variation in mRNA amounts can be detected.87
In the pathophysiology of intestinal stricture formation, a locally increased proliferation of mucosal myofibroblasts may occur. They exhibit a high proliferation rate coincident with the reduced migratory potential. Consequently, a local increase in cell number with reduced dispersing potential may lead to ineffective wound contraction and increased deposition of ECM, finally leading to tissue fibrosis (fig 8A, B). On the other hand, the reduced migratory potential of myofibroblasts from patients with Crohn’s disease could result in less migration of cells into the transmucosal wound areas, followed by proliferation of epithelial cells into these defects and finally the formation of fistulas (fig 8C). However, these models remain speculative, as the growth factor-induced migration of fibroblasts or the complex interaction of fibroblasts with immune cells or epithelial cells certainly have an important role also in wound healing, and no experimental data are available so far to decide which hypothesis is more reliable and promising.

Schematic representation of (A) normal wound healing, (B) fibrosis and (C) fistula formation, with effect on the role of fibroblasts. In the case of fibrosis, increased proliferation, migration and matrix synthesis occurs. In the pathogenesis of fistulas, reduced migration of myofibroblasts and synthesis of extracellular matrix is the problem. Epithelial cells try to close the wound area and migrate in the defect.
THE “ENCHAINING” END: INTESTINAL FIBROSIS
A chronic or recurrent inflammation causing chronic or recurrent tissue damage is considered a necessary condition for the initiation of intestinal fibrosis. Hypotheses for the development of strictures in IBD or other forms of enteritis often suggest an ongoing activation of collagen-producing myofibroblasts. In acute injury, the normal intestinal architecture is restored by post-transcriptional and post-translational mechanisms that prevent the net accumulation of ECM and fibrogenic cells. By contrast, during fibrosis, ECM accumulates and is not degraded at appropriate levels, whereas fibrogenic cells proliferate. Therefore, efforts should focus on exploring the mechanisms to prevent the activation of myofibroblasts.
The fibrotic mucosa of patients with IBD shows increased levels of mRNA and protein of collagen types I, III, IV and V.25,28,96,97 Which of these ECM proteins is predominant or most relevant to intestinal fibrosis is difficult to evaluate—until now, marked differences between Crohn’s disease or ulcerative colitis have not been proved.96 In other diseases associated with intestinal fibrosis—besides IBD, such as radiation enteritis—a similar ECM deposition pattern has been observed.1,2,98
TGF certainly has a role in the induction of collagen synthesis and matrix deposition.99 TGFβ is probably the most powerful and widely distributed pro-fibrogenic mediator in the body. It is essential for the physiological response to tissue injury, and is also involved in pathological fibrosis and fibrotic diseases of multiple tissues.100 Increased TGFβ levels for tissue fibrogenesis have a role in pulmonary fibrosis, liver fibrosis, chronic pancreatitis, scleroderma and renal glomerulosclerosis.100 Furthermore, a pathophysiological relevance was postulated for fibrotic complications of radiation therapy, chemotherapy and organ transplantation.101 The importance of TGFβ for fibrosis is indicated by increased protein expression in fibrotic tissue areas and also by the finding that administration of exogenous TGFβ to laboratory animals was followed by the development of fibrosis. Also, anti-TGFβ treatments reduce experimentally induced fibrosis.100
TGFβ alters the balance between ECM synthesis and degradation, inducing an increase in synthesis of matrix components and a decrease in ECM proteolytic activity, resulting in fibrogenesis and changes in the ECM composition. This has been extensively shown for liver fibrosis. TGFβ up regulates fibrillar and non-fibrillar collagens, fibronectin and tenascin, and the basement membrane proteins laminin and entactin.101 TGFβ also changes the splicing of fibronectin.100 A change in fibronectin splicing forms synthesised by intestinal myofibroblasts also occurs in IBD (unpublished data). Furthermore, TGFβ regulates the expression of TIMPs. Increased TIMP expression has been described in IBD.102
A relevant role of TGFβ has also been shown in experimental models of intestinal fibrosis.77,78 No good model for intestinal fibrosis is yet available to easily study pathophysiology and potential therapeutic strategies. The experimental models for studying TGF-mediated effects on intestinal fibrosis are specific, and TGF inhibition in models of severe colitis has been found to have disadvantages. This might be because TGFβ is also an important inductor of regulatory T cells. The inhibition of TGF may therefore reduce fibrosis but increase inflammation and tissue damage, which could be deleterious in the case of the intestinal wall. There are concerns that a general inhibition of TGF might be followed by insufficient wound healing, making unspecific anti-TGF treatment an unsuccessful approach. These concerns are even greater with respect to the intestinal wall and barrier. Impaired wound healing would cause persisting ulcers and tissue defects, making the barrier even more leaky than it is.
Another important growth factor implicated in intestinal fibrosis is IGF. Whereas IGF probably has beneficial effects and promotes mucosal repair and growth in the normal mucosa, evidence suggests that increased local IGFI expression may contribute to intestinal fibrosis.103 Increased IGFI expression is localised to myofibroblasts at sites of increased collagen mRNA expression and fibrosis in patients with Crohn’s disease and in animal models of chronic intestinal inflammation.104–107 IGFI stimulates collagen synthesis82,105,107,108 and it increases the proliferation of intestinal smooth-muscle cells and myofibroblasts.82,109–111 It also stimulates the migration of myofibroblasts.85 Endogenous TGFβ induces IGFI expression in intestinal fibroblasts, and subsequently IGFI stimulates the growth of these cells that have first been activated to a fibrogenic myofibroblast phenotype by TGF.82 Similarly, IGFII is a potent autocrine mitogen for intestinal fibroblasts.109
In addition to ECM synthesis, studies have been conducted on MMP expression as an important matrix-degrading system and its inhibitory counterpart TIMP in intestinal fibrosis. Interestingly, mesenchymal cells from the mucosa of patients with IBD showed increased levels and activity of MMP1, MMP2 and MMP3.25,112 Other reports indicate a normal expression of MMPs, with increased TIMP expression and activity.102 This was suggested as a potential mechanism for intestinal fibrosis and stricture formation, causing an accumulation of ECM via TIMP1-mediated inhibition of MMP activity.102 However, the data are not completely conclusive, and it is doubtful whether matrix deposition in intestinal fibrosis can be explained by an imbalance of the MMP–TIMP system.
Our knowledge of factors that normally interrupt or stop the process of intestinal fibrosis is limited. However, recent evidence suggests that suppressor of cytokine signalling (SOCS) proteins might be involved in limiting a fibrogenic tissue response. The SOCS proteins are a family of Src homology 2 domain-containing proteins. Currently, there are eight members of the SOCS family, of which some have been implicated in the negative regulation of signal transduction pathways induced by cytokines and growth factors.113 SOCS2 binds to type I IGFI receptor. In SOCS2-deficient mice, SOCS2 normally limits basal, IGFI-induced intestinal growth.114 Similarly, SOCS3 might be a mediator that limits growth factor action.115,116 Further studies will be required to show whether these findings lead to new therapeutic options.
There are already recent developments to therapeutically reduce fibrosis. A family of heparan sulphate mimetic polymers engineered to stimulate tissue repair, called ReGeneraTing Agents or RGTAs, was shown to induce ECM remodelling by decreasing collagen production.117 In biopsy specimens from patients with Crohn’s disease, ReGeneraTing Agents added in vitro decreased total collagen production by 50% and decreased collagen III synthesis by 76%.117
Further efforts and consecutive understanding of the pathophysiology of intestinal fibrosis will hopefully lead to new strategies and treatments that interrupt stricture formation, improving the outcome for patients who now have to undergo recurrent surgery.
SUMMARY
Although efforts for the development of anti-inflammatory treatments for patients with IBD have been extensive, treatments for fibrosis and fistulas in IBD are still lacking. As a first step, further studies are necessary to deepen our understanding of the mechanisms responsible for fibrosis in the mucosal (fig 9). This has to include the evaluation of inflammatory and anti-inflammatory mediators and their sources responsible for intestinal tissue damage, as well as mediators influencing the three main functions of mesenchymal cells in wound repair—migration, proliferation and ECM synthesis—in IBD in vitro and in vivo. This may lead to the development of treatments for modifying the wound-healing response and may offer advantages over current treatments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Potential strategies for future therapies. Present treatments mainly attempt to reduce inflammation. Attempts to intervene on different levels of fibrosis induction and development may prove to be favourable and beneficial in the future.
REFERENCES
Footnotes
-
Competing interests: None.